This post is just me amusing myself by exploring a tiny data set I have lying around. The dataset and the code is on Github.
In 2014 (I think), I was teaching the introductory cell biology labs (pictures in the linked post) in Linköping. We were doing a series of simple preparations to look at cells and organelles: a cheek swab gives you a view of dead mammalian cells with bacteria on them; Elodea gives you a nice chloroplast view; a red bell pepper gives you chromoplasts; and a banana stained with iodine gives you amyloplasts. Giving the same lab six times in a row, it became apparent how the number of stained amyloplasts decreased as the banana ripened.
I took one banana, sliced in into five pieces (named A-E), and left it out to ripen. Then I stained (with Lugol’s iodine solution) and counted the number of amyloplasts per cell in a few cells (scraped off with a toothpick) from the end of each piece at day 1, 5, and 9.
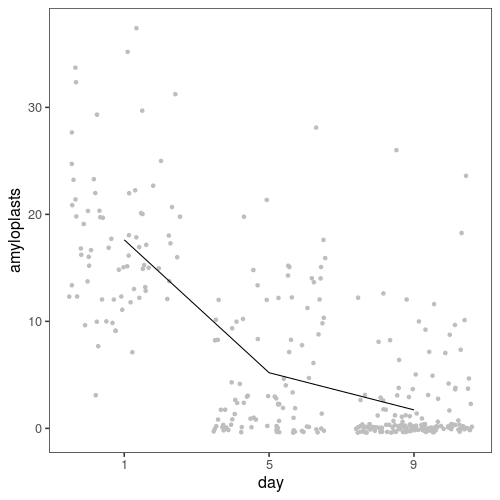
First, here is an overview of the data. On average, we go from 17 stained amyloplasts on day 1, to 5 on day five and 2 on day nine.
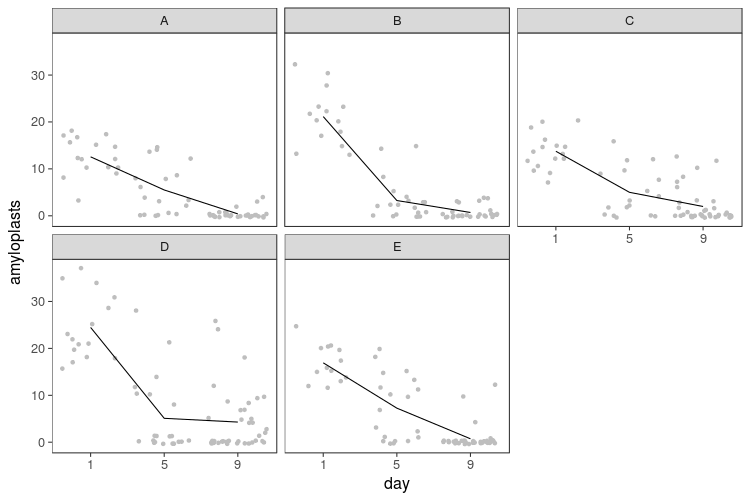
If we break the plot up by slices, we see decline in every slice and variability between them. Because I only sampled each slice once per day, there is no telling whether this is variation between parts of the banana or between samples taken (say, hypothetically, because I might have stuck the toothpick in more or less deeply, or because the ripeness varies from the middle to the peel).
How can we model this? Let’s first fit a linear model where the number of amyloplasts decline at a constant rate per day, allowing for different starting values and different declines for each slice. We can anticipate that a Gaussian linear model will have some problems in this situation.
We fit a linear model and pull out the fitted values for each day–slice combination:
model_lm <- lm(amyloplasts ~ day * slice,
data = banana)
levels <- expand.grid(slice = unique(banana$slice),
day = unique(banana$day),
stringsAsFactors = FALSE)
pred_lm <- cbind(levels,
predict(model_lm,
newdata = levels,
interval = "confidence"))
Then, to investigate the model’s behaviour, we can simulate data from the model, allowing for uncertainty in the fitted parameters, with the sim function from the arm package.
We make a function to simulate data from the linear model given a set of parameters, then simulate parameters and feed the first parameter combination to the function to get ourselves a simulated dataset.
y_rep_lm <- function(coef_lm, sigma, banana) {
slice_coef <- c(0, coef_lm[3:6])
names(slice_coef) <- c("A", "B", "C", "D", "E")
slice_by_day_coef <- c(0, coef_lm[7:10])
names(slice_by_day_coef) <- c("A", "B", "C", "D", "E")
banana$sim_amyloplasts <-
coef_lm[1] +
slice_coef[banana$slice] +
banana$day * (coef_lm[2] + slice_by_day_coef[banana$slice]) +
rnorm(nrow(banana), 0, sigma)
banana
}
sim_lm <- sim(model_lm)
sim_banana <- y_rep_lm(sim_lm@coef[1,], sim_lm@sigma[1], banana)
The result looks like this (black dots) compared with the real data (grey dots).
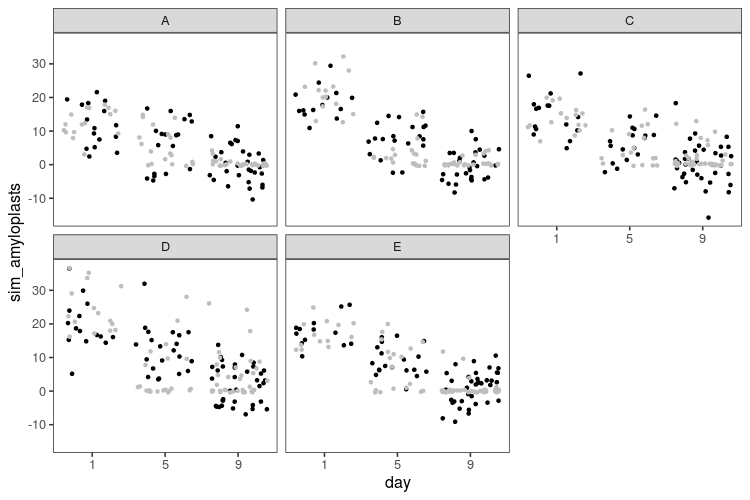
The linear model doesn’t know that the number of amyloplasts can’t go below zero, so it happily generates absurd negative values. While not apparent from the plots, the linear model also doesn’t know that amyloplasts counts are restricted to be whole numbers. Let’s fit a generalized linear model with a Poisson distribution, which should be more suited to this kind of discrete data. The log link function will also turn the linear decrease into an exponential decline, which seems appropriate for the decline in amyloplasts.
model_glm <- glm(amyloplasts ~ day * slice,
data = banana,
family = poisson(link = log))
pred_glm <- predict(model_glm,
newdata = levels,
se.fit = TRUE)
results_glm <- data.frame(levels,
average = pred_glm$fit,
se = pred_glm$se.fit,
stringsAsFactors = FALSE)
y_rep_glm <- function(coef_glm, banana) {
slice_coef <- c(0, coef_glm[3:6])
names(slice_coef) <- c("A", "B", "C", "D", "E")
slice_by_day_coef <- c(0, coef_glm[7:10])
names(slice_by_day_coef) <- c("A", "B", "C", "D", "E")
latent <- exp(coef_glm[1] +
slice_coef[banana$slice] +
banana$day * (coef_glm[2] + slice_by_day_coef[banana$slice]))
banana$sim_amyloplasts <- rpois(n = nrow(banana),
lambda = latent)
banana
}
sim_glm <- sim(model_glm)
sim_banana_glm <- y_rep_glm(sim_glm@coef[2,], banana)
This code is the same deal as above, with small modifications: glm instead of lm, with some differences in the interface. Then a function to simulate data from a Poisson model with an logarithmic link, that we apply to one set of parameters values.
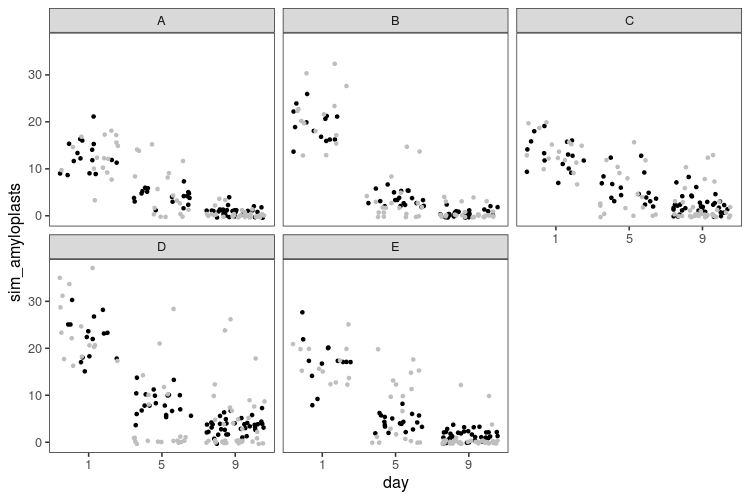
There are no impossible zeros anymore. However, there seems to be many more zeros in the real data than in the simulated data, and consequently, as the number of amyloplasts grow small, we overestimate how many there should be.
Another possibility among the standard arsenal of models is a generalised linear model with a negative binomial distribution. As opposed to the Poisson, this allows greater spread among the values. We can fit a negative binomial model with Stan.
library(rstan)
model_nb <- stan(file = "banana.stan",
data = list(n = nrow(banana),
n_slices = length(unique(banana$slice)),
n_days = length(unique(banana$day)),
amyloplasts = banana$amyloplasts,
day = banana$day - 1,
slice = as.numeric(factor(banana$slice)),
prior_phi_scale = 1))
y_rep <- rstan::extract(model_nb, pars = "y_rep")[[1]]
Here is the Stan code in banana.stan:
data {
int n;
int n_slices;
int <lower = 0> amyloplasts[n];
real <lower = 0> day[n];
int <lower = 1, upper = n_slices> slice[n];
real prior_phi_scale;
}
parameters {
real initial_amyloplasts[n_slices];
real decline[n_slices];
real < lower = 0> phi_rec;
}
model {
phi_rec ~ normal(0, 1);
for (i in 1:n) {
amyloplasts[i] ~ neg_binomial_2_log(initial_amyloplasts[slice[i]] +
day[i] * decline[slice[i]],
(1/phi_rec)^2);
}
}
generated quantities {
vector[n] y_rep;
for (i in 1:n) {
y_rep[i] = neg_binomial_2_rng(exp(initial_amyloplasts[slice[i]] +
day[i] * decline[slice[i]]),
(1/phi_rec)^2);
}
}
This model is similar to the Poisson model, except that the negative binomial allows an overdispersion parameter, a small value of which corresponds to large variance. Therefore, we put the prior on the reciprocal of the square root of the parameter.
Conveniently, Stan can also make the simulated replicated data for us in the generated quantities block.
What does the simulated data look like?
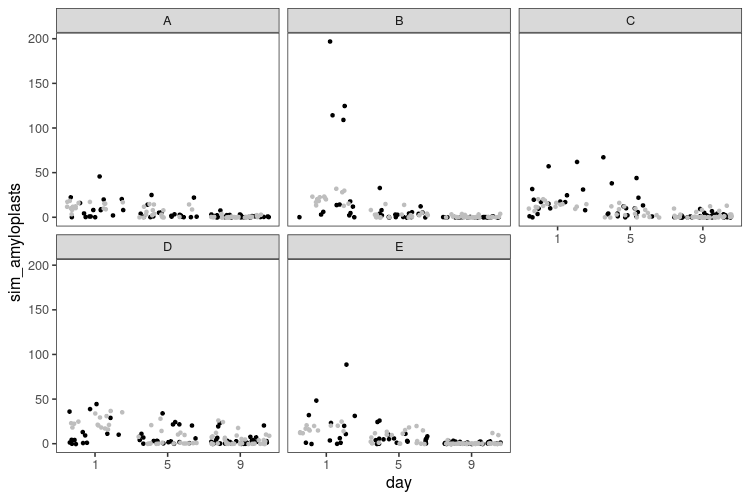
Here we have a model that allows for more spread, but in the process, generates some extreme data, with hundreds of amyloplasts per cell in some slices. We can try to be procrustean with the prior and constrain the overdispersion to smaller values instead:
model_nb2 <- stan(file = "banana.stan",
data = list(n = nrow(banana),
n_slices = length(unique(banana$slice)),
n_days = length(unique(banana$day)),
amyloplasts = banana$amyloplasts,
day = banana$day - 1,
slice = as.numeric(factor(banana$slice)),
prior_phi_scale = 0.1))
y_rep2 <- rstan::extract(model_nb2, pars = "y_rep")[[1]]
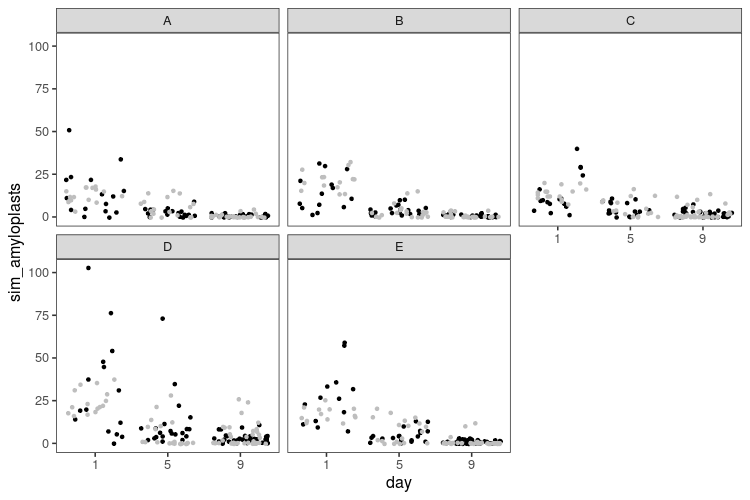
That looks a little better. Now, we’ve only looked at single simulated datasets, but we can get a better picture by looking at replicate simulations. We need some test statistics, so let us count how many zeroes there are in each dataset, what the maximum value is, and the sample variance, and then do some visual posterior predictive checks.
check_glm <- data.frame(n_zeros = numeric(1000),
max_value = numeric(1000),
variance = numeric(1000),
model = "Poisson",
stringsAsFactors = FALSE)
check_nb <- data.frame(n_zeros = numeric(1000),
max_value = numeric(1000),
variance = numeric(1000),
model = "Negative binomial",
stringsAsFactors = FALSE)
check_nb2 <- data.frame(n_zeros = numeric(1000),
max_value = numeric(1000),
variance = numeric(1000),
model = "Negative binomial 2",
stringsAsFactors = FALSE)
for (sim_ix in 1:1000) {
y_rep_data <- y_rep_glm(sim_glm@coef[sim_ix,], banana)
check_glm$n_zeros[sim_ix] <- sum(y_rep_data$sim_amyloplasts == 0)
check_glm$max_value[sim_ix] <- max(y_rep_data$sim_amyloplasts)
check_glm$variance[sim_ix] <- var(y_rep_data$sim_amyloplasts)
check_nb$n_zeros[sim_ix] <- sum(y_rep[sim_ix,] == 0)
check_nb$max_value[sim_ix] <- max(y_rep[sim_ix,])
check_nb$variance[sim_ix] <- var(y_rep[sim_ix,])
check_nb2$n_zeros[sim_ix] <- sum(y_rep2[sim_ix,] == 0)
check_nb2$max_value[sim_ix] <- max(y_rep2[sim_ix,])
check_nb2$variance[sim_ix] <- var(y_rep2[sim_ix,])
}
check <- rbind(check_glm,
check_nb,
check_nb2)
melted_check <- gather(check, "variable", "value", -model)
check_data <- data.frame(n_zeros = sum(banana$amyloplasts == 0),
max_value = max(banana$amyloplasts),
variance = var(banana$amyloplasts))
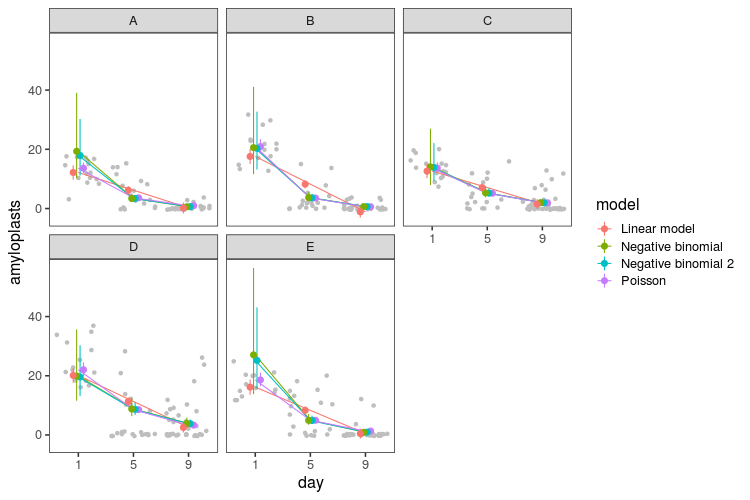
Here is the resulting distribution of these three discrepancy statistics in 1000 simulated datasets for the three models (generalised linear model with Poisson distribution and the two negative binomial models). The black line is the value for real data.
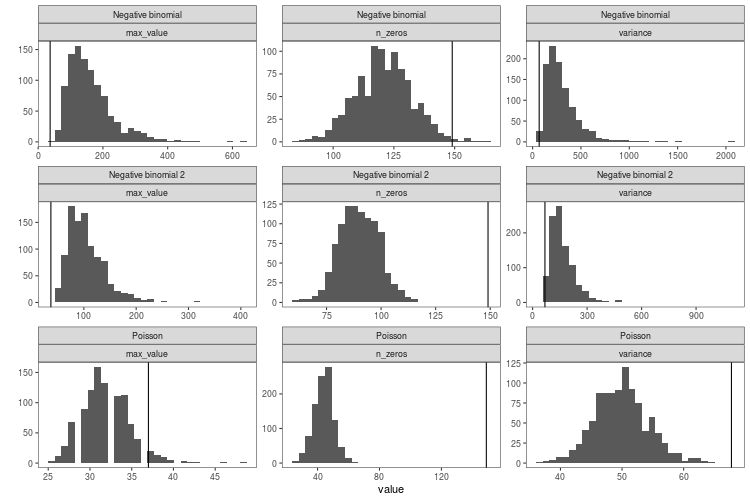
When viewed like this, it becomes apparent how the negative binomial models do not fit that well. The Poisson model struggles with the variance and the number of zeros. The negative binomial models get closer to the number of zeros in the real data, they still have too few, while at the same time having way too high maximum values and variance.
Finally, let’s look at the fitted means and intervals from all the models. We can use the predict function for the linear model and Poisson model, and for the negative binomial models, we can write our own:
pred_stan <- function(model, newdata) {
samples <- rstan::extract(model)
initial_amyloplasts <- data.frame(samples$initial_amyloplasts)
decline <- data.frame(samples$decline)
names(initial_amyloplasts) <- names(decline) <- c("A", "B", "C", "D", "E")
## Get posterior for levels
pred <- matrix(0,
ncol = nrow(newdata),
nrow = nrow(initial_amyloplasts))
for (obs in 1:ncol(pred)) {
pred[,obs] <- initial_amyloplasts[,newdata$slice[obs]] +
(newdata$day[obs] - 1) * decline[,newdata$slice[obs]]
}
## Get mean and interval
newdata$fit <- exp(colMeans(pred))
intervals <- lapply(data.frame(pred), quantile, probs = c(0.025, 0.975))
newdata$lwr <- exp(unlist(lapply(intervals, "[", 1)))
newdata$upr <- exp(unlist(lapply(intervals, "[", 2)))
newdata
}
pred_nb <- pred_stan(model_nb, levels)
pred_nb2 <- pred_stan(model_nb2, levels)
In summary, the three generalised linear models with log link function pretty much agree about the decline of amyloplasts during the later days, which looks more appropriate than a linear decline. They disagree about the uncertainty about the numbers on the first day, which is when there are a lot. Perhaps coincidentally, this must also be where the quality of my counts are the lowest, because it is hard to count amyloplasts on top of each other.