This is the second post about my journey towards writing more modern Tidyverse-style R code; here is the previous one. We will look at the common case of taking subset of data out of a data frame, making some complex R object from them, and then extracting summaries from those objects.
More nostalgia about plyr
I miss the plyr package. Especially ddply, ldply, and dlply, my favourite trio of R functions of all times. Yes, the contemporary Tidyverse package dplyr is fast and neat. And those plyr functions operating on arrays were maybe overkill; I seldom used them. But plyr was so smooth, so beautiful, and — after you’ve bashed your head against it for some time and it changed your mind — so intuitive. The package still exists, but it’s retired, and we shouldn’t keep writing R code like it’s 2009, should we?
I used to like to do something like this: take a data frame, push it through a function that returns some complex object, store those objects in a list, and then map various functions over that list to extract the parts I care about. What is the equivalent in the new idiom? To do the same thing but with the purrr package, of course! purrr replaces the list-centric parts of plyr, while dplyr covers data frame-centric summarisation, mutation and so on.
For this example we will be using the lm function on subsets of data and store the model object. It’s the simple case that everyone reaches for to demonstrate these features, but it’s a bit dubious. If you find yourself fitting a lot of linear models to subsets, maybe there are other models you should think about Especially here, when the fake data just happens to come from a multilevel model with varying intercepts … But in this case, let’s simulate a simple linear regression and look at the coefficients we get out.
set.seed(20210807)
n_groups <- 10
group_sizes <- rpois(n_groups, 10)
n <- sum(group_sizes)
fake_data <- tibble(id = 1:n,
group = rep(1:n_groups,
times = group_sizes),
predictor = runif(n, 0, 1))
group_intercept <- rnorm(n_groups, 0, 1)
fake_data$response <- fake_data$predictor * 10 +
group_intercept[fake_data$group] +
rnorm(n)
And here is the plyr code: First, dlply takes us from a data frame, splitting it by group, to a list of linear models. Then, ldply takes us from the list of models to a data frame of coefficients. tidy is a function from the wonderful broom package which extracts the same information as you would get in the rather unwieldy object from summary(lm), but as a data frame.
library(plyr)
library(broom)
fit_model <- function(data) {
lm(response ~ predictor, data)
}
models <- dlply(fake_data,
"group",
fit_model)
result <- ldply(models, tidy)
This is what the results might looks like. Notice how ldply adds the split labels nicely into the group column, so we know which rows came from which subset.
group term estimate std.error statistic p.value 1 1 (Intercept) -0.2519167 0.5757214 -0.4375670 6.732729e-01 2 1 predictor 10.6136902 1.0051490 10.5593207 5.645878e-06 3 2 (Intercept) 3.1528489 0.6365294 4.9531864 7.878498e-04 4 2 predictor 8.2075766 1.1458702 7.1627452 5.292586e-05 5 3 (Intercept) -0.8103777 0.6901212 -1.1742542 2.786901e-01 ...
split/map: The modern synthesis
If we pull out purrr, we can get the exact same table like so. The one difference is that we get a tibble (that is, a contemporary, more well-behaved data frame) out of it instead of a base R data.frame.
library(purrr)
models <- map(split(fake_data,
fake_data$group),
fit_model)
result <- map_df(models,
tidy,
.id = "group")
# A tibble: 80 x 6
group term estimate std.error statistic p.value
1 1 (Intercept) 1.67 0.773 2.16 6.32e- 2
2 1 predictor 8.67 1.36 6.39 2.12e- 4
3 2 (Intercept) 4.11 0.566 7.26 4.75e- 5
4 2 predictor 8.19 1.11 7.36 4.30e- 5
5 3 (Intercept) -7.50 0.952 -7.89 9.99e- 5
6 3 predictor 11.5 1.75 6.60 3.03e- 4
7 4 (Intercept) -19.8 0.540 -36.7 7.32e-13
8 4 predictor 11.5 0.896 12.8 5.90e- 8
9 5 (Intercept) -12.4 1.03 -12.0 7.51e- 7
10 5 predictor 9.69 1.82 5.34 4.71e- 4
# … with 70 more rows
First, the base function split lets us break the data into subsets based on the values of a variable, which in this case is our group variable. The output of this function is a list of data frames, one for each group.
Second, we use map to apply a function to each element of that list. The function is the same modelling function that we used above, which shoves the data into lm. We now have our list of linear models.
Third, we apply the tidy function to each element of that list of models. Because we want the result to be one single data frame consolidating the output from each element, we use map_df, which will combine the results for us. (If we’d just use map again, we would get a list of data frames.) The .id argument tells map to add the group column that indicates what element of the list of models each row comes from. We want this to be able to identify the coefficients.
If we want to be fancy, we can express with the Tidyverse-related pipe and dot notation:
library(magrittr) result <- fake_data %>% split(.$group) %>% map(fit_model) %>% map_df(tidy, .id = "group")
Nesting data into list columns
This (minus the pipes) is where I am at in most of my R code nowadays: split with split, apply with map and combine with map_dfr. This works well and looks neater than the lapply/Reduce solution discussed in part 0.
We can push it a step further, though. Why take the linear model out of the data frame, away from its group labels any potential group-level covariates — isn’t that just inviting some kind of mix-up of the elements? With list columns, we could store the groups in a data frame, keeping the data subsets and any R objects we generate from them together. (See Wickham’s & Grolemund’s R for Data Science for a deeper case study of this idea.)
library(dplyr)
library(tidyr)
fake_data_nested <- nest(group_by(fake_data, group),
data = c(id, predictor, response))
fake_data_models <- mutate(fake_data_nested,
model = map(data,
fit_model),
estimates = map(model,
tidy))
result <- unnest(fake_data_models, estimates)
First, we use the nest function to create a data frame where each row is a group, and the ”data” column contains the data for that subset.
Then, we add two list columns to that data frame: our list of models, and then our list of data frames with the coefficients.
Finally, we extract the estimates into a new data frame with unnest. The result is the same data frame of coefficients and statistics, also carrying along the data subsets, linear models and coefficents.
The same code with pipes:
fake_data %>%
group_by(group) %>%
nest(data = c(id, predictor, response)) %>%
mutate(model = map(data, fit_model),
estimates = map(model, tidy)) %>%
unnest(estimates) -> ## right way assignment just for kicks
result
I’m still a list column sceptic, but I have to concede that this is pretty elegant. It gets the job done, it keeps objects that belong together together so that there is no risk of messing up the order, and it is not that much more verbose. I especially like that we can run the models and extract the coefficients in the same mutate call.
Coda: mixed model
Finally, I can’t resist comparing the separate linear models to a linear mixed model of all the data.
We use lme4 to fit a varying-intercept model, a model that puts the same coefficient on the slope between the response and predictor in each group, but allows the intercepts to vary between groups, assuming them to be drawn from the same normal distribution. We put the coefficients from the linear models fit in each group separately and the linear mixed model together in the same data frame to be able to plot them.
library(ggplot2)
library(lme4)
model <- lmer(response ~ (1|group) + predictor,
fake_data)
lm_coef <- pivot_wider(result,
names_from = term,
values_from = estimate,
id_cols = group)
lmm_coef <- cbind(group = levels(model@flist$group),
coef(model)$group)
model_coef <- rbind(transform(lm_coef, model = "lm"),
transform(lmm_coef, model = "lmm"))
colnames(model_coef)[2] <- "intercept"
ggplot() +
geom_point(aes(x = predictor,
y = response,
colour = factor(group)),
data = fake_data) +
geom_abline(aes(slope = predictor,
intercept = intercept,
colour = factor(group),
linetype = model),
data = model_coef) +
theme_bw() +
theme(panel.grid = element_blank())
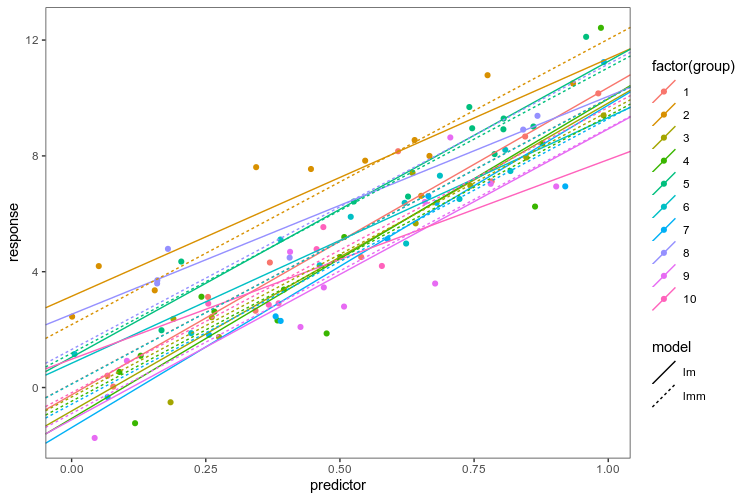
As advertised, the linear mixed model has the same slope in every group, and intercepts pulled closer to the mean. Now, we know that this is a good model for these fake data because we created them, and in the real world, that is obviously not the case … The point is: if we are going to fit a more complex model of the whole data, we want to be able to explore alternatives and plot them together with the data. Having elegant ways to transform data frames and summarise models at our disposal makes that process smoother.