Ken Richardson recently published an opinion piece about genetics titled ‘It’s the end of the gene as we know it‘. And I feel annoyed.
The overarching point of the piece is that there have been ‘radical revisions of the gene concept’ and that they ‘need to reach the general public soon—before past social policy mistakes are repeated’. He argues, among other things, that:
- headlines like ‘being rich and successful is in your DNA’ are silly;
- polygenic scores for complex traits have limited predictive power and problems with population structure;
- the classical concept of what a ‘gene’ has been undermined by molecular biology, which means that genetic mapping and genomic prediction are conceptually flawed.
You may be able to guess which of these arguments make me cheer and which make me annoyed.
There is a risk when you writes a long list of arguments, that if you make some good points and some weak points, no-one will remember anything but the weak point. Let us look at what I think are some good points, and the main weak one.
Gene-as-variant versus gene-as-sequence
I think Richardson is right that there is a difference in how classical genetics, including quantitative genetics, conceives of a ‘gene’, and what a gene is to molecular biology. This is the same distinction as Griffth & Stotz (2013), Portin & Wilkins (2017), and I’m sure many others have written about. (Personally, I used to call it ‘gene(1)’ and ‘gene(2)’, but that is useless; even I can’t keep track of which is supposed to be one and two. Thankfully, that terminology didn’t make it to the blog.)
In classical terms, the ‘gene’ is a unit of inheritance. It’s something that causes inherited differences between individuals, and it’s only observed indirectly through crosses and and differences between relatives. In molecular terms, a ‘gene’ is a piece of DNA that has a name and, optionally, some function. The these two things are not the same. The classical gene fulfills a different function in genetics than the molecular gene. Classical genes are explained by molecular mechanisms, but they are not reducible to molecular genes.
That is, you can’t just take statements in classical genetics and substitute ‘piece of DNA’ for ‘gene’ and expect to get anything meaningful. Unfortunately, this seems to be what Richardson wants to do, and this inability to appreciate classical genes for what they are is why the piece goes astray. But we’ll return to that in a minute.
A gene for hardwiring in your DNA
I also agree that a lot of the language that we use around genetics, casually and in the media, is inappropriate. Sometimes it’s silly (when reacting positively to animals, believing in God, or whatever is supposed to be ‘hard-wired in our DNA’) and sometimes it’s scary (like when a genetic variant was dubbed ‘The Warrior Gene’ on flimsy grounds and tied to speculations about Maori genetics). Even serious geneticists who should know better will put out press releases where this or that is ‘in your DNA’, and the literature is full of ‘genes for’ complex traits that have at best small effects. This is an area where both researchers and communicators should shape up.
Genomic prediction is hard
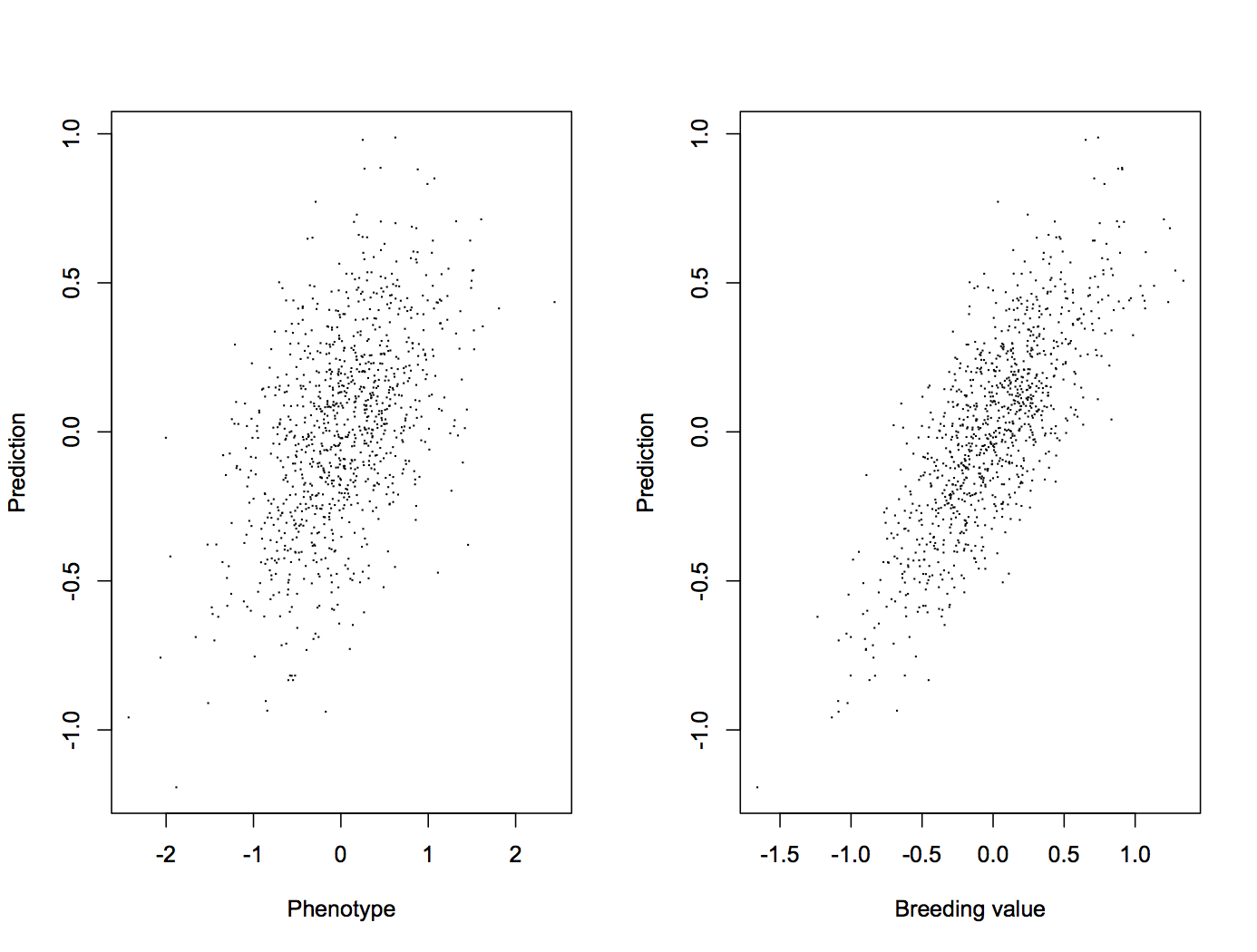
Polygenic scores are one form of genomic prediction, that is: one way to predict individuals’ trait values from their DNA. It goes something like this: you collect trait values and perform DNA tests on some reference population, then fit a statistical model that tells you which genetic variants differ between individuals with high and low trait values. Then you take that model and apply it to some other individuals, whose values you want to predict. There are a lot of different ways to do this, but they all amount to estimating how much each variant contributes to the trait, and somehow adding that up.
If you have had any exposure to animal breeding, you will recognise this as genomic selection, a technology that has been a boon to animal breeding in dairy cattle, pig, chicken, and to lesser extent other industries in the last ten years or so (see review by Georges, Charlier & Hayes 2018). It’s only natural that human medical geneticists want to do use the same idea to improve prediction of diseases. Unfortunately, it’s a bit harder to get genomic prediction to be useful for humans, for several reasons.
The piece touches on two important problems with genomic prediction in humans: First, DNA isn’t everything, so the polygenic scores will likely have to be combined with other risk factors in a joint model. It still seems to be an open question how useful genomic prediction will be for what diseases and in what contexts. Second, there are problems with population structure. Ken Richardson explains with an IQ example, but the broader point is that it is hard for the statistical models geneticists use to identify the causal effects in the flurry of spurious associations that are bound to exist in real data.
[A]ll modern societies have resulted from waves of migration by people whose genetic backgrounds are different in ways that are functionally irrelevant. Different waves have tended to enter the class structure at randomly different levels, creating what is called genetic population stratification. But different social classes also experience differences in learning opportunities, and much about the design of IQ tests, education, and so on, reflects those differences, irrespective of differences in learning ability as such. So some spurious correlations are, again, inevitable.
So, it may be really hard to get good genomic predictors that predict accurately. This is especially pressing for studies of adaptation, where researchers might use polygenic scores estimated in European populations to compare other populations, for example. Methods to get good estimates in the face of population structure is a big research topic in both human, animal, and plant genetics. I wouldn’t be surprised if good genomic prediction in humans would require both new method development and big genome-wide association studies that cover people from all of the world.
These problems are empirical research problems. Polygenic scores may be useful or not. They will probably need huge studies with lots of participants and new methods with smart statistical tricks. However, they are not threatened by conceptual problems with the word ‘gene’.
Richardson’s criticism is timely. We’d all like to think that anyone who uses polygenic scores would be responsible, pay attention to the literature about sensitivity to population structure, and not try to over-interpret average polygenic scores as some way to detect genetic differences between populations. But just the other week, an evolutionary psychology journal published a paper that did just that. There are ill-intentioned researchers around, and they enjoy wielding the credibility of fancy-sounding modern methods like polygenic scores.
Genetic variants can be causal, though
Now on to where I think the piece goes astray. Here is a description of genetic causation and how that is more complicated than it first seems:
Of course, it’s easy to see how the impression of direct genetic instructions arose. Parents “pass on” their physical characteristics up to a point: hair and eye color, height, facial features, and so on; things that ”run in the family.” And there are hundreds of diseases statistically associated with mutations to single genes. Known for decades, these surely reflect inherited codes pre-determining development and individual differences?
But it’s not so simple. Consider Mendel’s sweet peas. Some flowers were either purple or white, and patterns of inheritance seemed to reflect variation in a single ”hereditary unit,” as mentioned above. It is not dependent on a single gene, however. The statistical relation obscures several streams of chemical synthesis of the dye (anthocyanin), controlled and regulated by the cell as a whole, including the products of many genes. A tiny alteration in one component (a ”transcription factor”) disrupts this orchestration. In its absence the flower is white.
So far so good. This is one of the central ideas of quantitative genetics: most traits that we care about are complex, in that an individual’s trait value is affected by lots of genes of individually small effects, and to a large extent on environmental factors (that are presumably also many and subtle in their individual effects). Even relatively simple traits tend to be more complicated when you look closely. For example, almost none of the popular textbook examples of single gene traits in humans are truly influenced by variants at only one gene (Myths of human genetics). Most of the time they’re either unstudied or more complicated than that. And even Mendelian rare genetic diseases are often collections of different mutations in different genes that have similar effects.
This is what quantitative geneticists have been saying since the early 1900s (setting aside the details about the transcription factors, which is interesting in its own right, but not a crucial part of the quantitative genetic account). This is why genome-wide association studies and polygenic scores are useful, and why single-gene studies of ‘candidate genes’ picked based on their a priori plausible function is a thing of the past. But let’s continue:
This is a good illustration of what Noble calls ”passive causation.” A similar perspective applies to many ”genetic diseases,” as well as what runs in families. But more evolved functions—and associated diseases—depend upon the vast regulatory networks mentioned above, and thousands of genes. Far from acting as single-minded executives, genes are typically flanked, on the DNA sequence, by a dozen or more ”regulatory” sequences used by wider cell signals and their dynamics to control genetic transcription.
This is where it happens. We get a straw biochemist’s view of the molecular gene, where everything is due only to protein-coding genes that encode one single protein that has one single function, and then he enumerates a lot of different exceptions to this view that is supposed to make us reject the gene concept: regulatory DNA (as in the quote above), dynamic gene regulation during development, alternative splicing that allows the same gene to make multiple protein isoforms, noncoding RNA genes that act without being turned into protein, somatic rearrangements in DNA, and even that similar genes may perform different functions in different species … However, the classical concept of a gene used in quantitative genetics is not the same as the molecular gene. Just because the molecular biology and classical genetics both use the word ‘gene’, users of genome-wide association studies are not forced to commit to any particular view about alternative splicing.
It is true that there are ‘vast regulatory networks’ and interplay at the level of ‘the cell as a whole’, but that does not prevent some (or many) of the genes involved in the network to be affected by genetic variants that cause differences between the individuals. That builds up to form genetic effects on traits, through pathways that are genuinely causal, ‘passive’ or not. There are many genetic variants and complicated indirect mechanisms involved. The causal variants are notoriously hard to find. They are still genuine causes. You can become a bit taller because you had great nutrition as a child rather than poor nutrition. You can become a bit taller because you carry certain genetic variants rather than others.