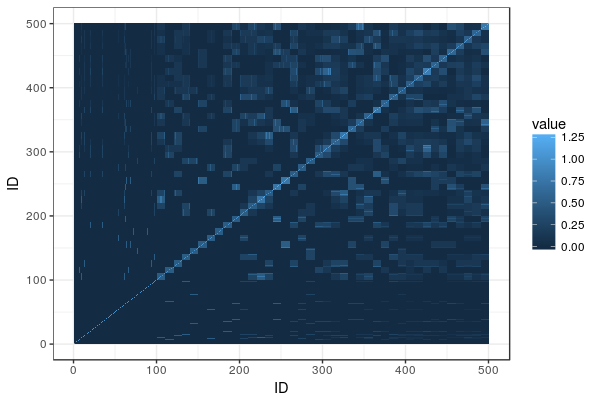
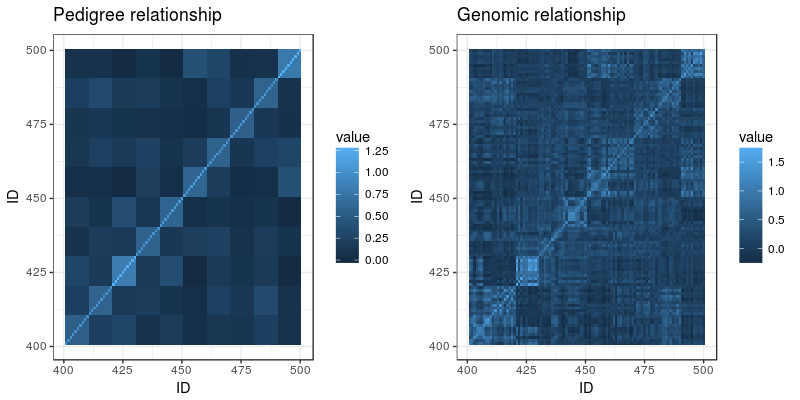
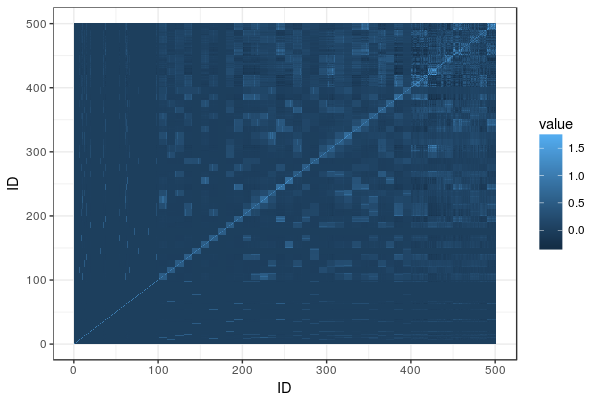
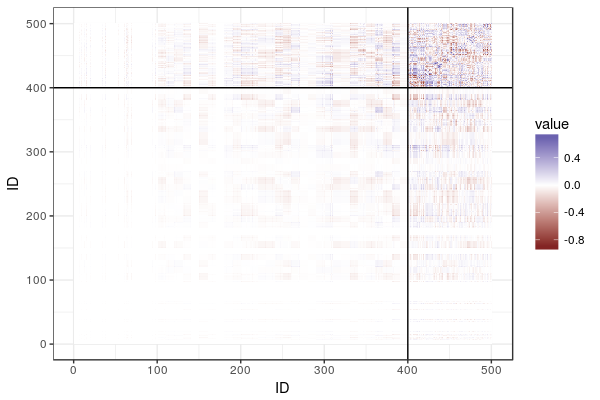
A lot of dairy cattle is crossbred, but genomic evaluation is often done within breed. What about the crossbred individuals? This paper (VanRaden et al. 2020) describes the US Council on Dairy Cattle Breeding’s crossbred genomic prediction that started 2019.
In short, the method goes like this: They describe each crossbred individual in terms of their ”genomic breed composition”, get predictions for each them based on models from all the breeds separately, and then combine the results in proportion to the genomic breed composition. The paper describes how they estimate the genomic breed composition, and evaluated accuracy by predicting held-out new data from older data.
The genomic breed composition is a delightfully elegant hack: They treat ”how much breed X is this animal” as a series of traits and run a genomic evaluation on them. The training set: individuals from sets of reference breeds with their trait value set to 100% for the breed they belong to and 0% for other breeds. ”Marker effects for GBC [genomic breed composition] were then estimated using the same software as for all other traits.” Neat. After some adjustment, they can be interpreted as breed percentages, called ”base breed representation”.
As they already run genomic evaluations from each breed, they can take these marker effects and then animal’s genotypes, and get one estimate for each breed. Then they combine them, weighting by the base breed representation.
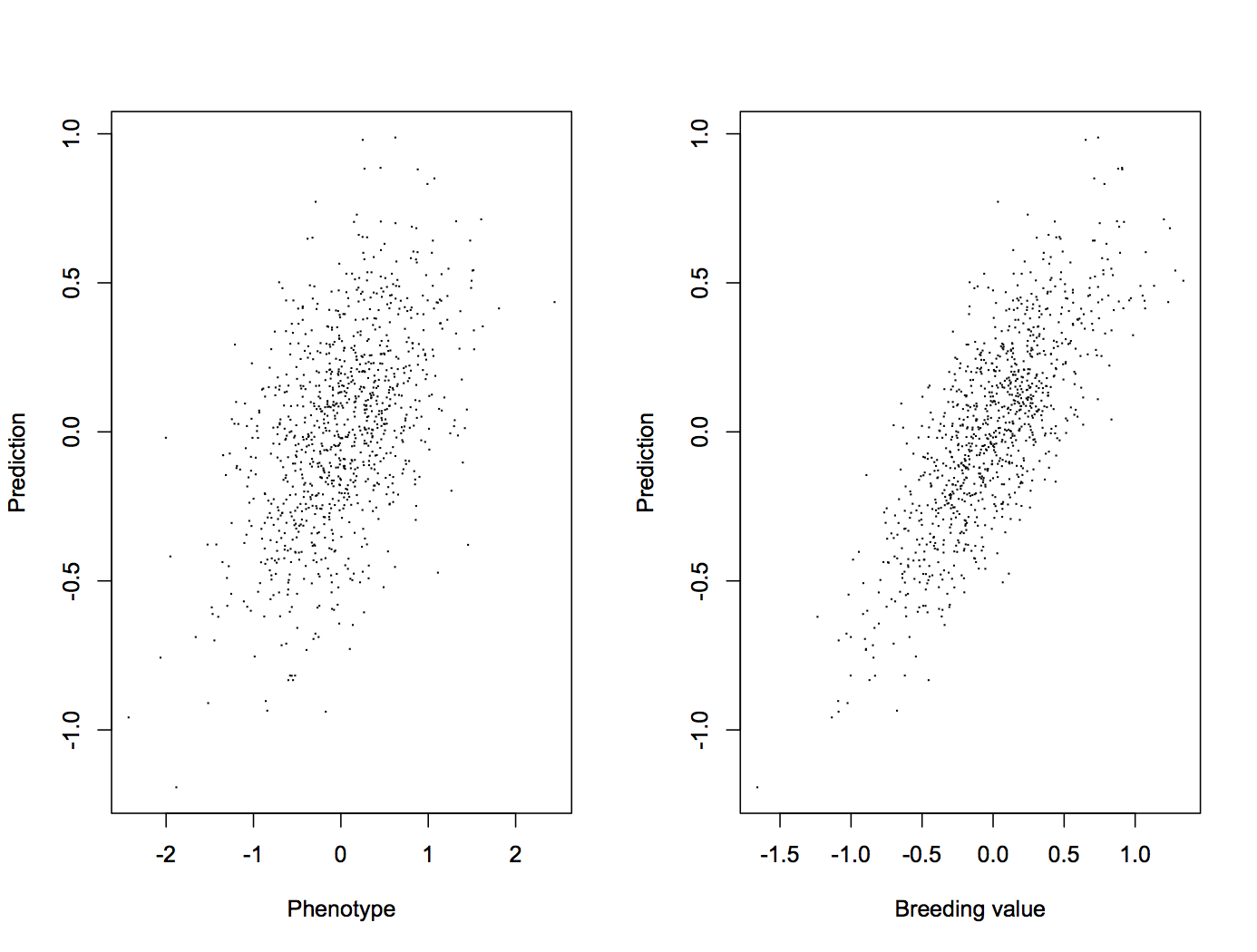
Does it work? Yes, in the sense that it provides genomic estimates for animals that otherwise wouldn’t have any, and that it beats parent average estimates.
Accuracy of GPTA was higher than that of [parent average] for crossbred cows using truncated data from 2012 to predict later phenotypes in 2016 for all traits except productive life. Separate regressions for the 3 BBR categories of crossbreds suggest that the methods perform equally well at 50% BBR, 75% BBR, and 90% BBR.
They mention in passing comparing these estimates to estimates from a common set of marker effects for all breeds, but there is no detail about that model or how it compared in accuracy.
The discussion starts with this sentence:
More breeders now genotype their whole herds and may expect evaluations for all genotyped animals in the future.
That sounds like a reasonable expectation, doesn’t it? Before what they could do with crossbred genotypes was to throw it away. There are lots of other things that might be possible with crossbred evaluation in the future (pulling in crossbred data into the evaluation itself, accounting for ancestry in different parts of the genome, estimating breed-of-origin of alleles, looking at dominance etc etc).
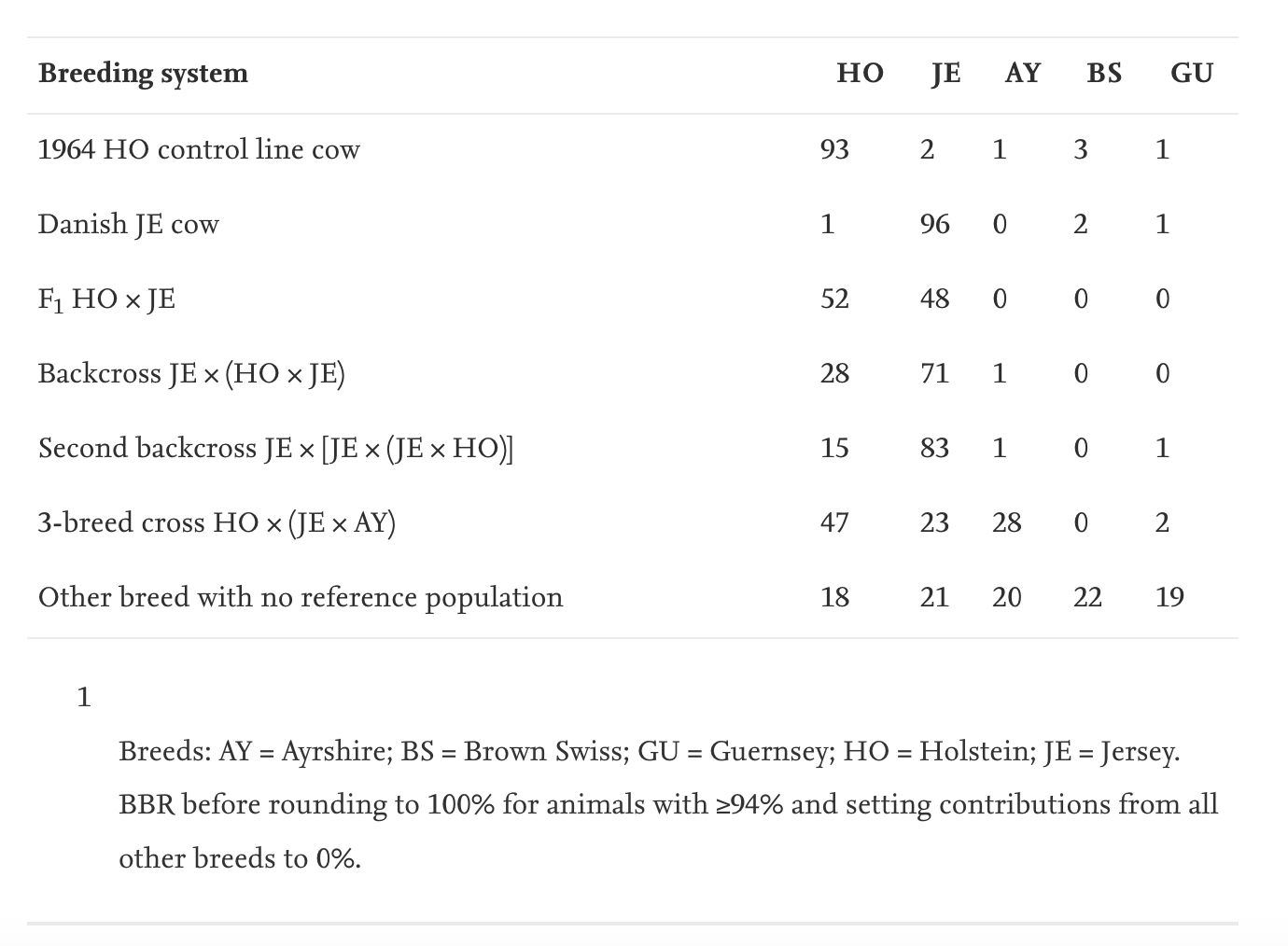
My favourite result in the paper is Table 8, which shows:
Example BBR for animals from different breeding systems are shown in Table 8. The HO cow from a 1964 control line had 1960s genetics from a University of Minnesota experimental selection project and a relatively low relationship to the current HO population because of changes in breed allele frequencies over the past half-century. The Danish JE cow has alleles that differ somewhat from the North American JE population. Other examples in the table show various breed crosses, and the example for an animal from a breed with no reference population shows that genetic contributions from some other breed may be evenly distributed among the included breeds so that BBR percentages sum to 100. These examples illustrate that GBC can be very effective at detecting significant percentages of DNA contributed by another breed.
Literature
VanRaden, P. M., et al. ”Genomic predictions for crossbred dairy cattle.” Journal of Dairy Science 103.2 (2020): 1620-1631.