(Here is a a paper from a recent journal club.)
Nasser et al. (2021) present a way to prioritise potential noncoding causative variants. This is part of solving the fine mapping problem, i.e. how to find the underlying causative variants from genetic mapping studies. They do it by connecting regulatory elements to genes and overlapping those regulatory elements with variants from statistical fine-mapping. Intuitively, it might not seem like connecting regulatory elements to genes should be that hard, but it is a tricky problem. Enhancers — because that is the regulatory element most of this is about; silencers and insulators get less attention — do not carry any sequence that tells us what gene they will act on. Instead, this needs to be measured or predicted somehow. These authors go the measurement route, combining chromatin sequencing with chromosome conformation capture.
This figure from the supplementary materials show what the method is about:

Additional figure 1 from Nasser et al. (2021) showing an overview of the workflow and an example of two sets of candidate variants derived from-fine mapping, each with variants that overlap enhancers connected to IL10.
They use chromatin sequence data (such as ATAC-seq, histone ChIP-seq or DNAse-seq) in combination with HiC chromosome conformation capture data to identify enhancers and connect them to genes (this was developed earlier in Fulco et al. 2019). The ”activity-by-contact model” means to multiply the contact frequency (from HiC) between promoter and enhancer with the enhancer activity (read counts from chromatin sequencing), standardised by the total contact–activity product with all elements within a window. Fulco et al. (2019) previously found that this conceptually simple model did well at connecting enhancers to genes (as evaluated with a CRISPR inhibition assay called CRISPRi-FlowFISH, which we’re not going into now, but it’s pretty ingenious).
In order to use this for fine-mapping, they calculated activity-by-contact maps for every gene combined with every open chromatin element within 5 Mbp for 131 samples from ENCODE and other sources. The HiC data were averaged of contacts in ten cell types, transformed to be follow a power-law distribution. That is, they did not do the HiC analysis per cell type, but use the same average HiC contact matrix combined with chromatin data from different cell types. Thus, the specificity comes from the promoters and enhancers that are called as active in each sample — I assume this is because the HiC data comes from a different (and smaller) set of cell types than the chromatin sequencing. Element–gene pairs that reached above a threshold were considered connected, for a total of about six million connections, involving 23,000 genes and 270,000 enhancers. On average, a gene had 2.8 enhancers and an enhancer connected to 2.7 genes.
They picked putative causative variants by overlapping the variant sets with these activity-by-contact maps and selecting the highest scoring enhancer gene pair.They used fine-mapping results from multiple previous studies. These variants were estimated with different methods, but they are all some flavour of fine-mapping by variable selection. Statistical fine mapping estimate sets of variants, called credible sets, that have high posterior probability of being the causative variant. They included only completely noncoding credible sets, i.e. those that did not include a coding sequence or splice variant. They applied this to 72 traits in humans, generating predictions for ~ 5000 noncoding credible sets of variants.
Did it work?
Variants for fine-mapping were enriched in connected enhancers more than in open chromatin in general, in cell types that are relevant to the traits. In particular, inflammatory bowel disease variants were enriched in enhancers in 65 samples, including immune cell types and gut cells. The strongest enrichment was in activated dendritic cells.
They used a set of genes previously known to be involved in inflammatory bowel disease, assuming that they were the true causative genes for their respective noncoding credible sets, and then compared their activity-by-contact based prioritisation of the causative gene to simply picking the closest gene. Picking the closest gene was right in 30 out of 37 sets. Picking the gene with the highest activity-by-contact score was right in 17 cases out of 18 sets that overlapped an activity-by-contact enhancer. Thus, this method had higher precision but worse recall. They also tested a number of eQTL-based, enrichment and enhancer–gene mapping methods, that did worse.
What it tells us about causative variants
Most of the putative causative variants, picked based on maximising activity-by-contact, were close to the proposed gene (median 13 kbp) and most involved the closest gene (77%). They were often found their putative causative variants to be located in enhancers that were only active in a few cell- or tissue types (median 4), compared to the promoters of the target genes, that were active in a broader set (median 120). For example, in inflammatory bowel disease, there were several examples where the putatively causal enhancer was only active in a particular immune cell or a stimulated state of an immune cell.
My thoughts
What I like about this model is that it is so different to many of the integrative and machine learning methods we see in genomics. It uses two kinds of data, and relatively simple computations. There is no machine learning. There is no sequence evolution or conservation analysis. Instead, measure two relevant quantities, standardise and preprocess them a bit, and then multiply them together.
If the success of the activity-by-contact model for prioritising causal enhancers generalises beyond the 18 causative genes investigated in the original paper, this is an argument for simple biology-based heuristics over machine learning models. It also suggest that, in the absence of contact data, one might do well by prioritising variant in enhancers that are highly active in relevant cell types, and picking the closest gene as the proposed causative gene.
However, the dataset needs to cover the relevant cell types, and possibly cells that are in the relevant stimulated states, meaning that it provides a motivation for rich conditional atlas-style datasets of chromatin and chromosome conformation.
I am, personally, a little bit sad that expression QTL methods seem to doing poorly. On the other hand, it makes some sense that eQTL might not have the genomic resolution to connect enhancers to genes.
Finally, if the relatively simple activity-by-contact model or the ridiculously simple method of ”picking the closest gene” beats machine learning models using the same data types and more, it suggests that the machine learning methods might not be solving theright problem. After all, they are not trained directly to prioritise variants for complex traits — because there are too few known causative variants for complex traits.
Literature
Fulco, C. P., Nasser, J., Jones, T. R., Munson, G., Bergman, D. T., Subramanian, V., … & Engreitz, J. M. (2019). Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nature genetics.
Nasser, J., Bergman, D. T., Fulco, C. P., Guckelberger, P., Doughty, B. R., Patwardhan, T. A., … & Engreitz, J. M. (2021). Genome-wide enhancer maps link risk variants to disease genes. Nature.
 , trait 2 only
, trait 2 only  , or both traits
, or both traits  , and are the defaults good?
, and are the defaults good?  ) … This means that you can look at single trait association data, and get an idea of the number of marginal associations, possibly dependent on allele frequency. The estimates from a gene expression dataset and a genome-wide association catalog work out to a prior around
) … This means that you can look at single trait association data, and get an idea of the number of marginal associations, possibly dependent on allele frequency. The estimates from a gene expression dataset and a genome-wide association catalog work out to a prior around  , which is the coloc default. So far so good.
, which is the coloc default. So far so good.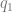 and
and 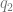 . But not all of the genome is functional. If you could straightforwardly define a functional proportion, you could just divide by it.
. But not all of the genome is functional. If you could straightforwardly define a functional proportion, you could just divide by it.