We have a new preprint posted to Biorxiv looking into the genetic basis of bone strength and other bone properties in crossbred laying hens in two different housing environments (furnished cages and floor pens).
Here are the citation and abstract:
Martin Johnsson, Helena Wall, Fernando A Lopes Pinto, Robert H. Fleming, Heather A. McCormack, Cristina Benavides-Reyes, Nazaret Dominguez-Gasca, Estefania Sanchez-Rodriguez, Ian C. Dunn, Alejandro B. Rodriguez-Navarro, Andreas Kindmark, Dirk-Jan de Koning (2021) Genetics of tibia bone properties of crossbred commercial laying hens in different housing systems. bioRxiv 2021.06.21.449243
Osteoporosis and bone fractures are a severe problem for the welfare of laying hens, with genetics and environment, such as housing system, each making substantial contributions to bone strength. In this work, we performed genetic analyses of bone strength, bone mineral density and bone composition, as well as body weight, in 860 commercial crossbred laying hens from two different companies, kept in either furnished cages or floor pens. We compared bone traits between housing systems and crossbreds, and performed a genome-wide association study of bone properties and body weight.
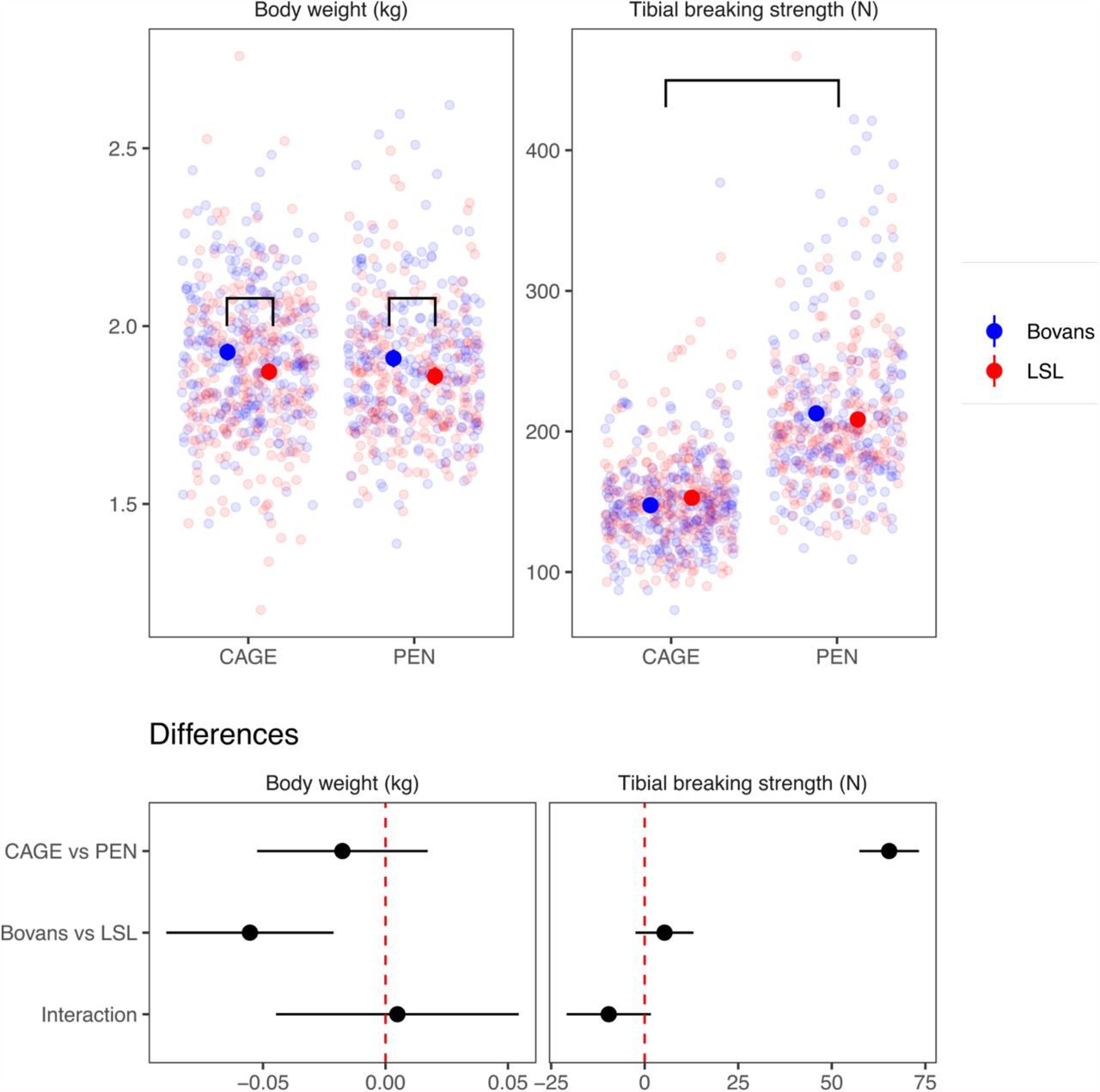
As expected, the two housing systems produced a large difference in bone strength, with layers housed in floor pens having stronger bones. These differences were accompanied by differences in bone geometry, mineralisation and chemical composition. Genome-scans either combining or independently analysing the two housing systems revealed no genome-wide significant loci for bone breaking strength. We detected three loci for body weight that were shared between the housing systems on chromosomes 4, 6 and 27 (either genome-wide significant or suggestive when the housing systems were analysed individually) and these coincide with associations for bone length.
In summary, we found substantial differences in bone strength, content and composition between hens kept in floor pens and furnished cages that could be attributed to greater physical activity in pen housing. We found little evidence for large-effect loci for bone strength in commercial crossbred hens, consistent with a highly polygenic architecture for bone strength in the production environment. The lack of consistent genetic associations between housing systems in combination with the differences in bone phenotypes support gene-by-environment interactions with housing system.
The background is that bone quality is a serious problem for laying hens; that housing systems that allow for more movement are known to lead to stronger bones; and that previous work on the genetics of bone parameters comes mostly from pure lines or from experimental intercrosses between divergent lines. Here, we study commercial crossbred laying hens from two different companies.
Being housed in a floor pen, where there is more opportunity for physical activity, or in a furnished cage makes a big difference to bone breaking strength. For comparison, we also show body weight, which is not much different between the housing environments. This difference was accompanied by differences in bone composition (see details in the paper).
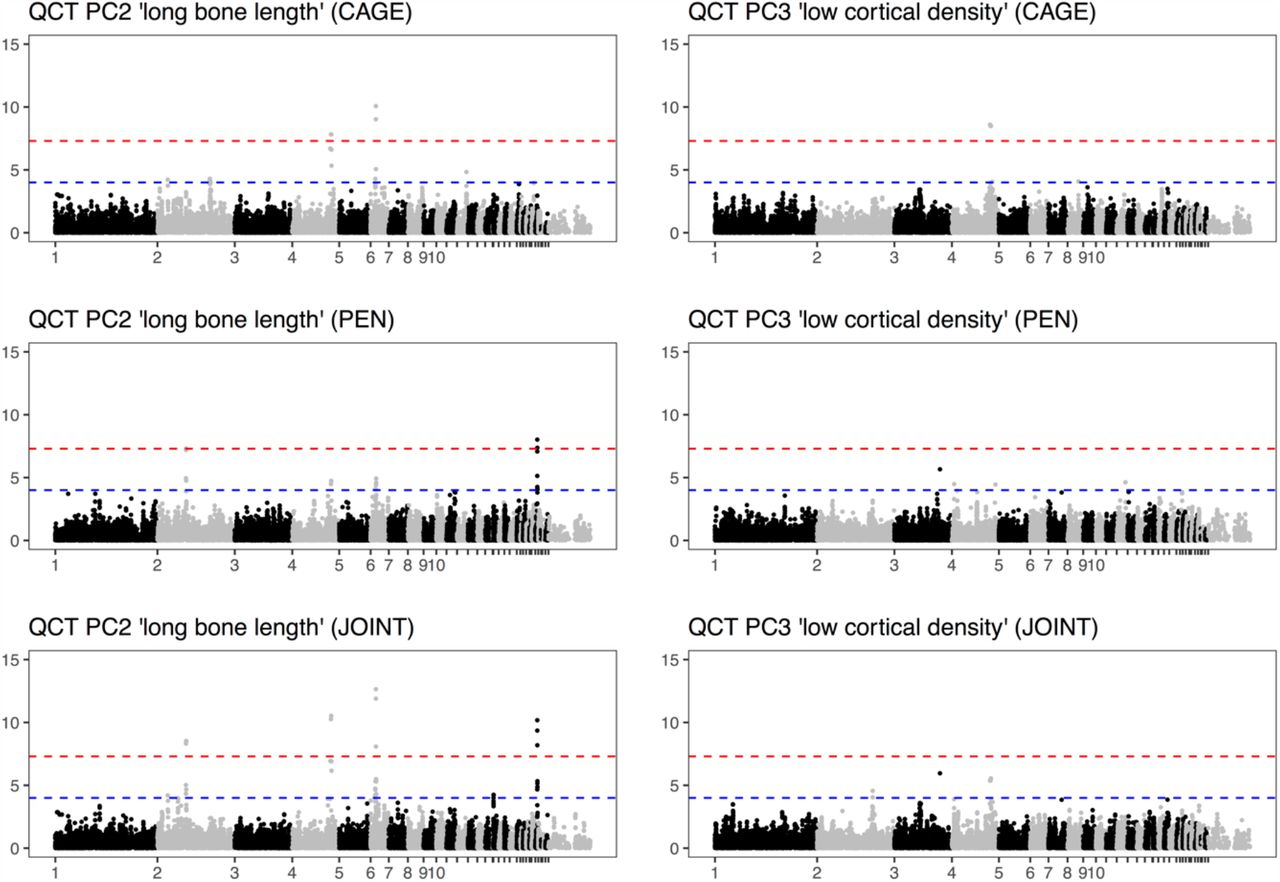
And here are the Manhattan plots from genome-wide association: bone strength shows no major loci, as opposed to body weight, which has strong associations that are shared between the housing systems.
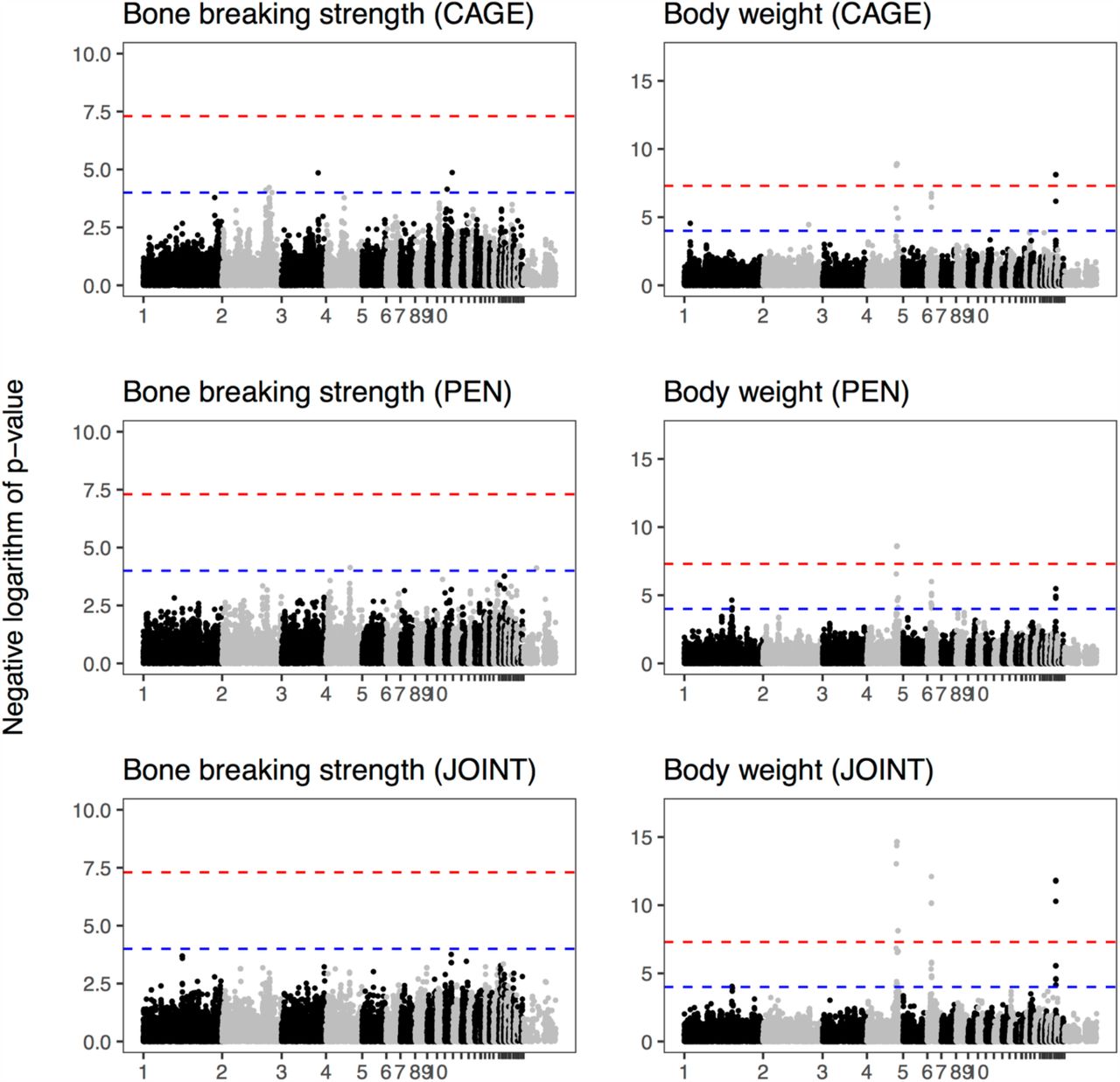
And if we compare the genome-wide associations, marker for marker, between the housing systems, there is nothing in common between the suggestive associations for bone strength. (Body weight below for comparison.)

This includes not detecting major loci for bone strength that have been found in pure lines of chickens. We think this is due to gene-by-environment interactions with housing (i.e. physical activity). This might be a complication for genomic selection for bone quality, as selection might need to be targeted to different housing systems.
Finally, the three strong association for body weight shown above overlap previously detected loci on chromosomes 4, 6, and 27. We do not have the genomic resolution to nominate candidate genes with any confidence, but the chromosome 4 locus overlaps both the CCKAR gene, which is a strong candidate for growth and body mass in the chicken and the LCORL/NCAPG locus, which has been associated with body size in several species. These regions (plus a fourth one) are also associated with bone length: