The other day was the first
Virtual breeding and genetics journal club organised by John Cole. This was the first online journal club I’ve attended (shocking, given how many video calls I’ve been on for other sciencey reasons), so I thought I’d write a little about it: both the format and the paper. You can look the
slide deck from the journal club here (pptx file).
The medium
We used Zoom, and that seemed to work, as I’m sure anything else would, if everyone just mute their microphone when they aren’t speaking. As John said, the key feature of Zoom seems to be the ability for the host to mute everyone else. During the call, I think we were at most 29 or so people, but only a handful spoke. It will probably get more intense with the turn taking if more people want to speak.
The format
John started the journal club with a code of conduct, which I expect helped to set what I felt was a good atmosphere. In most journal clubs I’ve been in, I feel like the atmosphere has been pretty good, but I think we’ve all heard stories about hyper-critical and hostile journal clubs, and that doesn’t sound particularly fun or useful. On that note, one of the authors, Oscar González-Recio, was on the call and answered some questions.
The paper
Saborío‐Montero, Alejandro, et al. ”
Structural equation models to disentangle the biological relationship between microbiota and complex traits: Methane production in dairy cattle as a case of study.” Journal of Animal Breeding and Genetics 137.1 (2020): 36-48.
The authors measured methane emissions (by analysing breath with with an infrared gas monitor) and abundance of different microbes in the rumen (with Nanopore sequencing) from dairy cows. They genotyped the animals for relatedness.
They analysed the genetic relationship between breath methane and abundance of each taxon of microbe, individually, with either:
- a bivariate animal model;
- a structural equations model that allows for a causal effect of abundance on methane, capturing the assumption that the abundance of a taxon can affect the methane emission, but not the other way around.
They used them to estimate heritabilities of abundances and genetic correlations between methane and abundances, and in the case of the structural model: conditional on the assumed causal model, the effect of that taxon’s abundance on methane.
My thoughts
It’s cool how there’s a literature building up on genetic influences on the microbiome, with some consistency across studies. These intense high-tech studies on relatively few cattle might build up to finding new traits and proxies that can go into larger scale phenotyping for breeding.
As the title suggests, the paper advocates for using the structural equations model: ”Genetic correlation estimates revealed differences according to the usage of non‐recursive and recursive models, with a more biologically supported result for the recursive model estimation.” (Conclusions)
While I agree that a priori, it makes sense to assume a structural equations model with a causal structure, I don’t think the results provide much evidence that it’s better. The estimates of heritabilities and genetic correlations from the two models are near indistinguishable. Here is the key figure 4, comparing genetic correlation estimates:
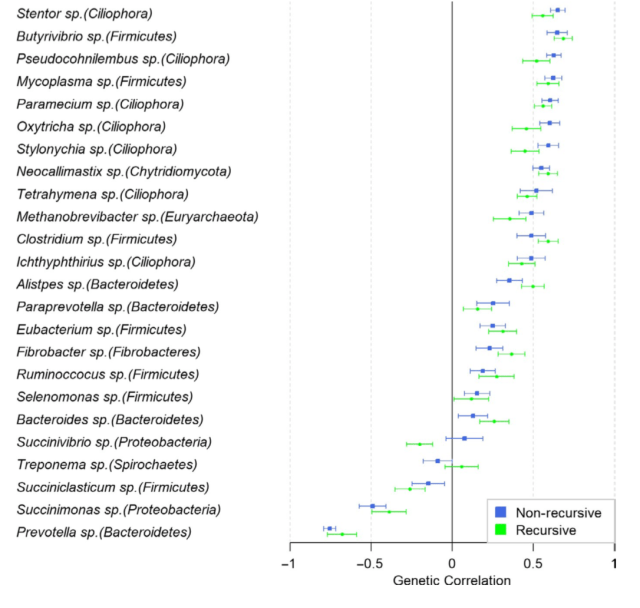
As you can see, there are a couple of examples of genetic correlations where the point estimate switches sign, and one of them (
Succinivibrio sp.) where the credible intervals don’t overlap. ”Recursive” is the structural equations model. The error bars are 95% credible intervals. This is not strong evidence of anything; the authors are responsible about it and don’t go into interpreting this difference. But let us speculate! They write:
All genera in this case, excepting Succinivibrio sp. from the Proteobacteria phylum, resulted in overlapped genetic cor- relations between the non‐recursive bivariate model and the recursive model. However, high differences were observed. Succinivibrio sp. showed the largest disagreement changing from positively correlated (0.08) in the non‐recursive bivariate model to negatively correlated (−0.20) in the recursive model.
Succinivibrio are also the taxon with the estimated largest inhibitory effect on methane (from the structural equations model).
While some taxa, such as ciliate protozoa or Methanobrevibacter sp., increased the CH4 emissions …, others such as Succinivibrio sp. from Proteobacteria phylum decreased it
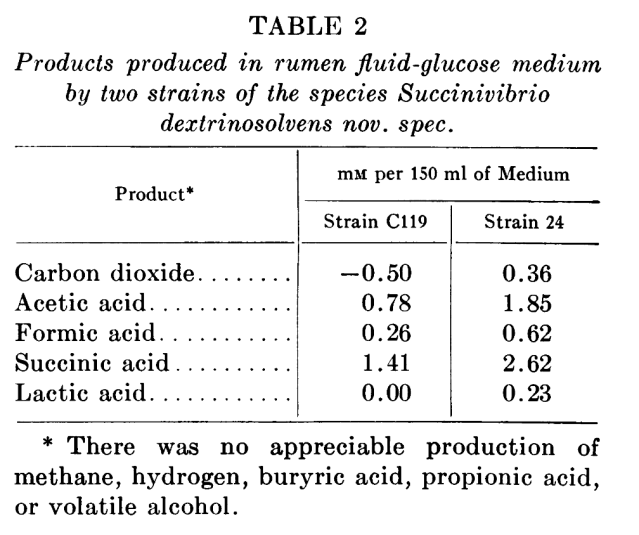
Looking at the paper that first described these bacteria (Bryan & Small 1955),
Succinivibrio were originally isolated from the cattle rumen, and their name is because ”they ferment glucose with the production of a large amount of succinic acid”. Bryant & Small made a fermentation experiment to see what came out, and it seems that the bacteria don’t produce methane:
This is also in line with a rRNA sequencing study of high and low methane emitting cows (
Wallace & al 2015) that found lower
Succinivibrio abundance in high methane emitters.
We may speculate that
Succinivibrio species could be involved in diverting energy from methanogens, and thus reducing methane emissions. If that is true, then the structural equations model estimate (larger genetic negative correlation between
Succinivibrio abundance and methane) might be better than one from the animal model.
Finally, while I’m on board with the a priori argument for using a structural equations model, as with other applications of causal modelling (gene networks, Mendelian randomisation etc), it might be dangerous to consider only parts of the system independently, where the microbes are likely to have causal effects on each other.
Literature
Saborío‐Montero, Alejandro, et al. ”
Structural equation models to disentangle the biological relationship between microbiota and complex traits: Methane production in dairy cattle as a case of study.” Journal of Animal Breeding and Genetics 137.1 (2020): 36-48.
Wallace, R. John, et al. ”
The rumen microbial metagenome associated with high methane production in cattle.”
BMC genomics 16.1 (2015): 839.
Bryant, Marvin P., and Nola Small. ”
Characteristics of two new genera of anaerobic curved rods isolated from the rumen of cattle.”
Journal of bacteriology 72.1 (1956): 22.