A few weeks ago I posted about fitting the quantitative genetic animal model with MCMCglmm and R-INLA. Since then, I listened to a talk by Lars Rönnegård, one of the creators of the hglm package, and this paper was published in GSE about animal models in Stan.
hglm
The hglm package fits hierarchical generalised linear models. That includes the animal model with pedigree or genomic relatedness. Hierarchical generalised linear models also allow you to model the dispersion of random effects, which lets you do tricks like variance QTL mapping (Rönnegård & Valdar 2011), breeding values for variances (Rönnegård et al. 2010) or genomic prediction models with predictors of marker variance (Mouresan, Selle & Rönnegård 2019). But let’s not get ahead of ourselves. How do we fit an animal model?
Here is the matrix formulation of the animal model that we skim through in every paper. It’s in this post because we will use the design matrix interface to hglm, which needs us to give it these matrices (this is not a paper, so we’re not legally obliged to include it):

The terms are the the trait value, intercept, fixed coefficients and their design matrix, genetic coefficients and their design matrix, and the residual. The design matrix Z will contain one row and column for each individual, with a 1 to indicate its position in the phenotype table and pedigree and the rest zeros. If we sort our files, it’s an identity matrix.
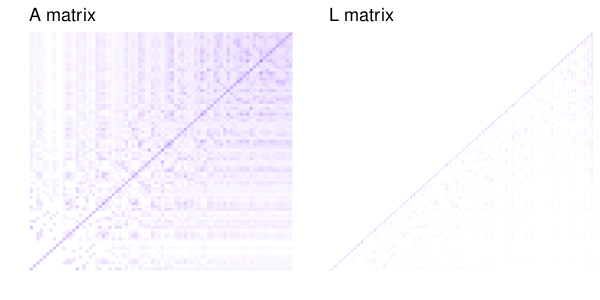
The trick with the genetic coefficients is that they’re correlated, with a specific known correlation structure that we know from the pedigree (or in genomic models, from markers). It turns out (Lee, Nelder & Pawitan 2017, chapter 8) that you can change the Z matrix around so that it lets you fit the model with an identity covariance matrix, while still accounting for the correlations between relatives. You replace the random effects for relatedness with some transformed random effects that capture the same structure. One way to do this is with Cholesky decomposition.

As an example of what the Cholesky decomposition does, here is slice of the additive relationship matrix of 100 simulated individuals (the last generation of one replicate of these simulations) and the resulting matrix from Cholesky decomposition.
So instead of
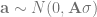
We can fit
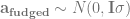
This lets us fit the animal model with hglm, by putting in a modified Z matrix.
Assuming we have data frames with a pedigree and a phenotype (like, again, from these simulations):
library(AGHmatrix)
library(hglm)
A <- Amatrix(ped)
Z0 <- diag(1000)
L <- t(chol(A))
Z <- Z0 %*% L
X <- model.matrix(~1, pheno)
model <- hglm(y = pheno$pheno,
X = X,
Z = Z,
conv = 1e-8)
est_h2 <- model$varRanef / (model$varRanef + model$varFix)
(I found the recommendation to decrease the convergence criterion from the default for animal models in a YouTube video by Xia Chen.)
Stan
When we turn to Stan, we will meet the Cholesky trick again. Stan is a software for Markov Chain Monte Carlo, built to fit hierarchical linear models, and related high-dimensional models, more effectively than other sampling strategies (like Gibbs). rstan is a helpful package for running Stan from within R.
Nishio & Arakawa (2019) recently published a Stan script to fit an animal model, comparing Stan to a Gibbs sampler (and a related MCMC sampler that they also didn’t publish the code for). If we look into their Stan model code, they also do a Cholesky decomposition to be able to use an identity matrix for the variance.
First, they decompose the additive relationship matrix that the program takes in:
transformed data{
matrix[K,K] LA;
LA = cholesky_decompose(A);
}
And then, they express the model like this:
vector[N] mu; vector[K] a; a_decompose ~ normal(0, 1); a = sigma_G * (LA * a_decompose); mu = X * b + Z * a; Y ~ normal(mu, sigma_R);
We can add this line to the generated quantities block of the Stan program to get heritability estimates directly:
real h2; h2 = sigma_U / (sigma_U + sigma_E)
Here, we’ve saved their model to a stan file, and now we can run it from R:
pheno$scaled_pheno <- as.vector(scale(pheno$pheno))
model_stan <- stan(file = "nishio_arakawa.stan",
data = list(Y = pheno$scaled_pheno,
X = X,
A = A,
Z = Z0,
J = 1,
K = 1000,
N = 1000))
est_h2_stan <- summary(model_stan, pars = "h2")$summary
Important note that I always forget: It's important to scale your traits before you run this model. If not, the priors might be all wrong.
The last line pulls out the summary for the heritability parameter (that we added above). This gives us an estimate and an interval.
The paper also contains this entertaining passage about performance, which reads as if it was a response to a comment, actual or anticipated:
R language is highly extensible and provides a myriad of statistical and graphical techniques. However, R language has poor computation time compared to Fortran, which is especially well suited to numeric computation and scientific computing. In the present study, we developed the programs for GS and HMC in R but did not examine computation time; instead, we focused on examining the performance of estimating genetic parameters and breeding values.
Yes, two of their samplers (Gibbs and HMC) were written in R, but the one they end up advocating (and the one used above), is in Stan. Stan code gets translated into C++ and then compiled to machine code.
Stan with brms
If rstan lets us run Stan code from R and examine the output, brms lets us write down models in relatively straightforward R syntax. It’s like the MCMCglmm of the Stan world. We can fit an animal model with brms too, by directly plugging in the relationship matrix:
model_brms <- brm(scaled_pheno ~ 1 + (1|animal),
data = pheno,
family = gaussian(),
cov_ranef = list(animal = A),
chains = 4,
cores = 1,
iter = 2000)
Then, we can pull out the posterior samples for the variability, here expressed as standard errors, compute the heritability and then get the estimates (and interval, if we want):
posterior_brms <- posterior_samples(model_brms,
pars = c("sd_animal", "sigma"))
h2_brms <- posterior_brms[,1]^2 /
(posterior_brms[,1]^2 + posterior_brms[,2]^2)
est_h2_brms <- mean(h2_brms)
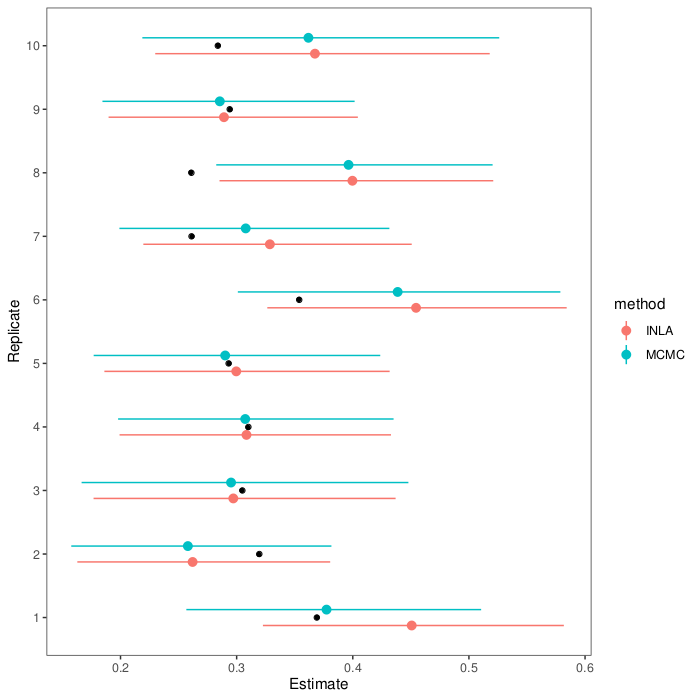
(Code is on GitHub: both for the graphs above, and the models.)