Our paper on genetic variation in recombination in the pig just came out the other week. I posted about it already when it was a preprint, but we dug a little deeper into some of the results in response to peer review, so let us have a look at it again.
Summary
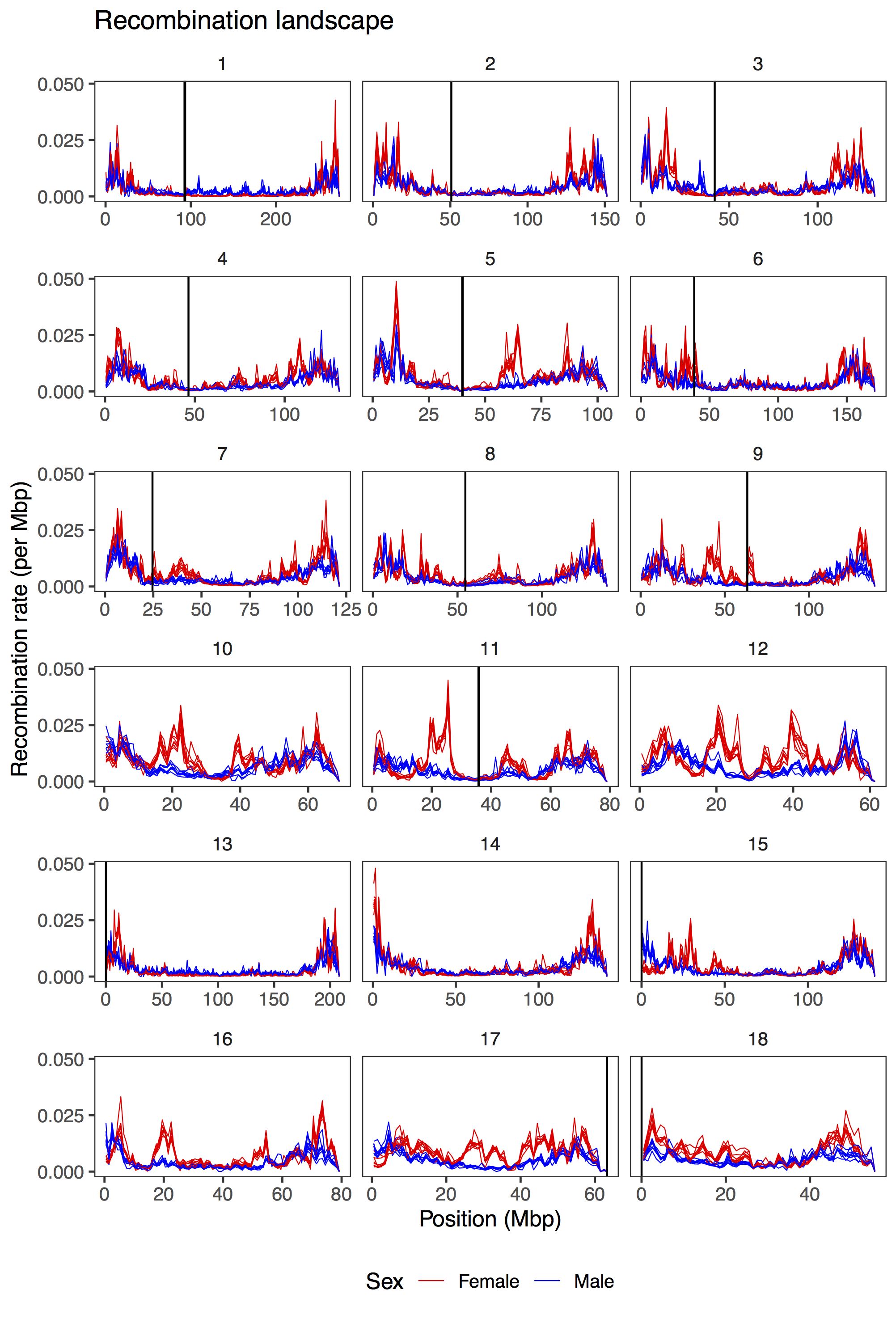
Recombination between chromosomes during meiosis leads to shuffling of genetic material between chromosomes, creating new combinations of alleles. Recombination rate varies, though, between parts of the genome, between sexes, and between individuals. Illustrating that, here is a figure from the paper showing how recombination rate varies along the chromosomes of the pig genome. Female recombination rate is higher than male recombination rate on most chromosomes, and in particular in regions of higher recombination rate in the middle of certain chromosomes.
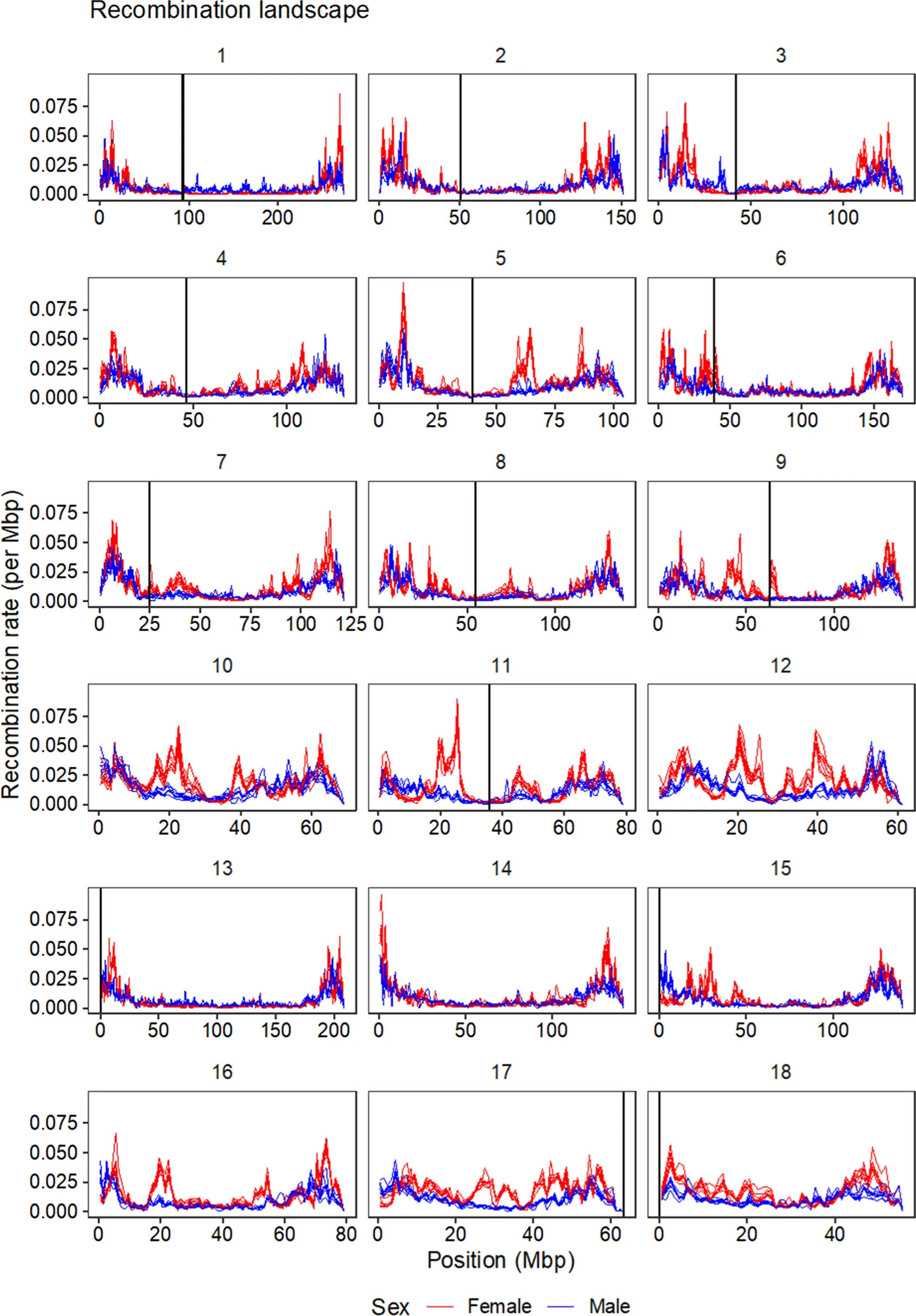
Fig 2 from the paper: average recombination rate in 1 Mbp windows along the pig genome. Each line is a population, coloured by sexes.
In several other vertebrates, part of that individual variation in recombination rate (in the gametes passed on by that individual) is genetic, and associated with regions close to known meiosis-genes. It turns out that this is the case in the pig too.
In this paper, we estimated recombination rates in nine genotyped pedigree populations of pigs, and used that to perform genome-wide association studies of recombination rate. The heritability of autosomal recombination rate was around 0.07 in females, 0.05 in males.
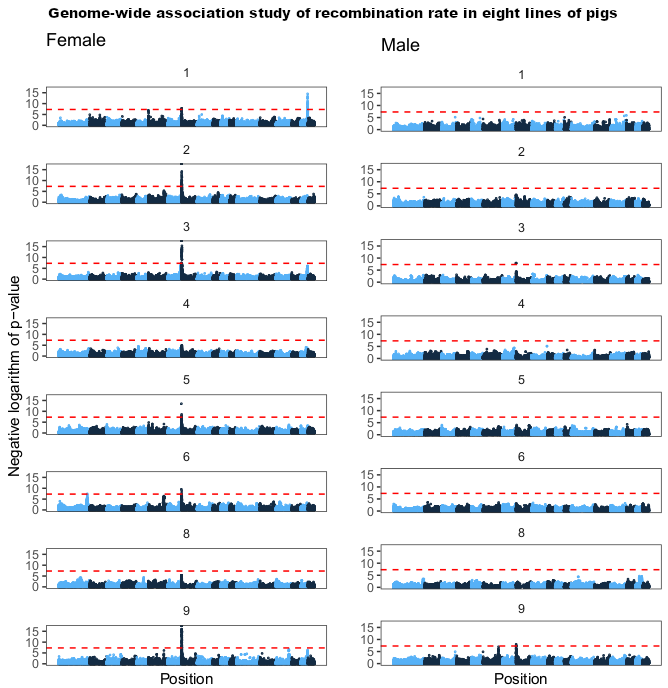
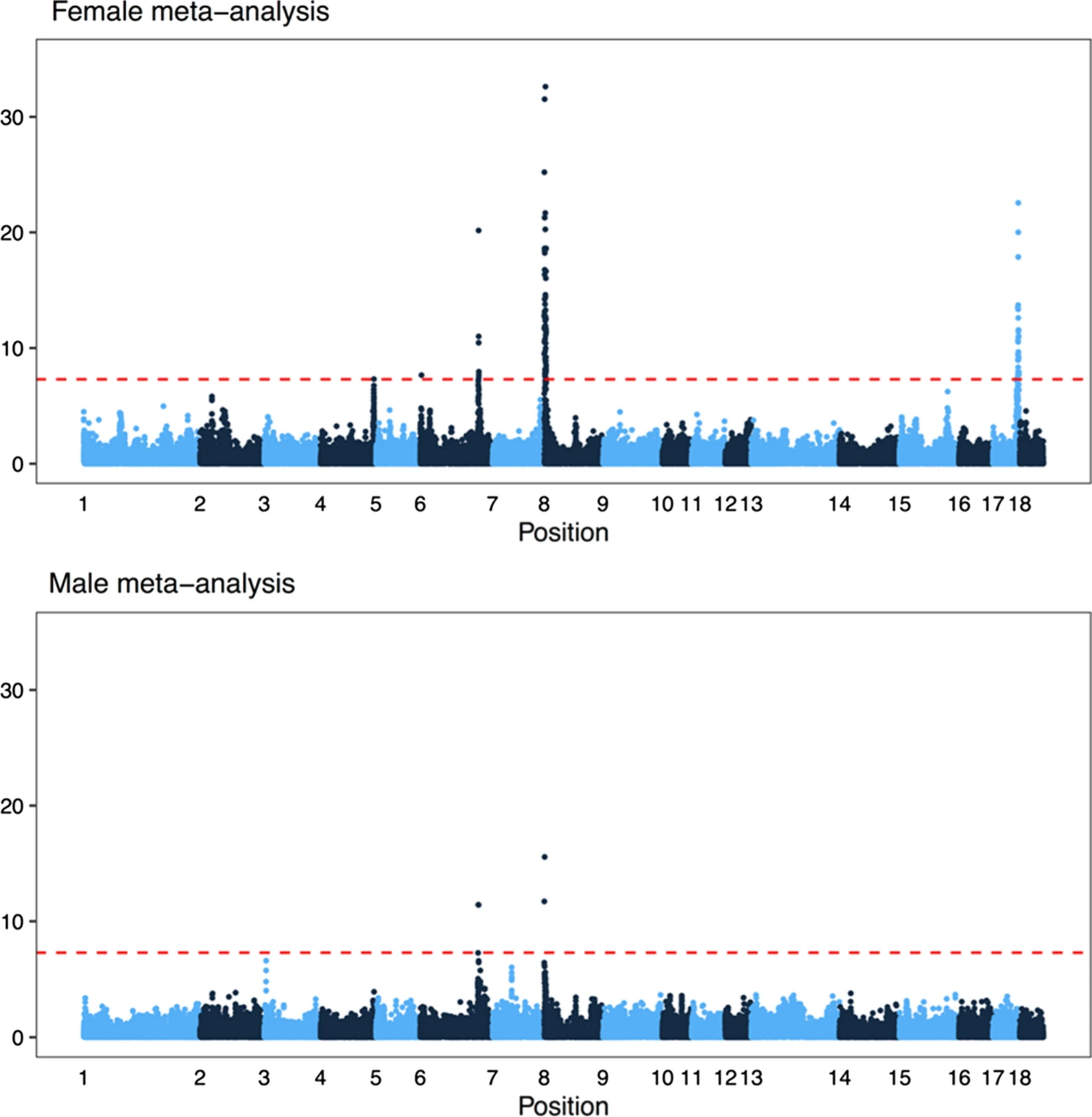
The major genome-wide association hit on chromosome 8, well known from other mammals, overlapped the RNF212 gene in most populations in females, and to a lesser extent in males.

Fig 6 from the paper, showing genome-wide association results for eight of the populations (one had too few individuals with recombination rate estimates after filtering for GWAS). The x-axis is position in the genome, and the y-axis is the negative logarithm of the p-value from a linear mixed model with repeated measures.
One of the things we added after the preprint is a meta-analysis of genome-wide association over all the populations (separated by sex). In total, there were six associated regions, five of which are close to known recombination genes: RNF212, SHOC1, SYCP2, MSH4 and HFM1. In particular, several of the candidates are genes involved in whether a double strand break resolves as a crossover or non-crossover. However, we do not have the genomic resolution to know whether these are actually the causative genes; there are significant markers overlapping many genes, and the candidate genes are not always the closest gene.
How well does the recombination landscape agree with previous maps?
The recombination landscapes accord pretty well with Tortereau et al.’s (2012) maps. We find a similar sex difference, with higher recombination in females on all autosomes except chromosome 1 and 13, and a stronger association with GC content in females. However, our recombination rates tend to be higher, possibly due to some overestimation. Different populations, where recombinations are estimated independently, also have similar recombination landscapes.
But it doesn’t agree so well with Lozada-Soto et al. (2021), does it?
No, that is true. Between preprint and finished version, Lozada-Soto et al. (2021) published a genome-wide association study of recombination rate in the pig. They found heritabilities of recombination rate of a similar magnitude as we did, but their genome-wide association results are completely different. We did not find the hits that they found, and they did not find the hits we found, or any previously known candidate genes for recombination. To be honest, I don’t have a good explanation for these differences.
How about recombination hotspots and PRDM9?
At a very fine scale, most recombination tends to occur in hotspots of around a few kilobasepairs. As this study used pedigrees and SNP chips with much coarser density than this, we cannot say much about the fine-scale recombination landscape. We work, at the finest, with windows of 1 Mbp. However, as the pig appears to have a working and rapidly evolving PRDM9 gene (encoding a protein that is responsible for recombination hotspot targeting), the pig probably has a PRDM9-based landscape of hotspots just like humans and mice (Baker et al. 2019).
Tortereau et al. (2012) found a positive correlation between counts of the PRDM9 DNA-binding motif and recombination rate, which is biologically plausible, as more PRDM9 motifs should mean more hotspots. So, we estimated this correlation for comparison, but found only a very weak relationship — this is one point where our results are inconsistent with previous maps. This might be because of changes in improved pig genome assembly we use, or it might be an indication that we have worse genomic resolution due to the genotype imputation involved in our estimation. However, one probably shouldn’t expect to find strong relationships between a process at the kilobasepair-scale when using windows of 1 Mbp in the first place.
Can one breed for increased recombination to improve genetic gain?
Not really. Because recombination breaks up linkage disequlibrium between causative variants, higher recombination rate could reveal genetic variation for selection and improve genetic gain. However, previous studies suggest that recombination rate needs to increase quite a lot (two-fold or more) to substantially improve breeding (Battagin et al. 2016). We made some back of the envelope quantitative genetic calculations on the response to selection for recombination, and it would be much smaller than that.
Literature
Johnsson M*, Whalen A*, Ros-Freixedes R, Gorjanc G, Chen C-Y, Herring WO, de Koning D-J, Hickey JM. (2021) Genetics variation in recombination rate in the pig. Genetics Selection Evolution (* equal contribution)
Tortereau, F., Servin, B., Frantz, L., Megens, H. J., Milan, D., Rohrer, G., … & Groenen, M. A. (2012). A high density recombination map of the pig reveals a correlation between sex-specific recombination and GC content. BMC genomics, 13(1), 1-12.
Lozada‐Soto, E. A., Maltecca, C., Wackel, H., Flowers, W., Gray, K., He, Y., … & Tiezzi, F. (2021). Evidence for recombination variability in purebred swine populations. Journal of Animal Breeding and Genetics, 138(2), 259-273.
Baker, Z., Schumer, M., Haba, Y., Bashkirova, L., Holland, C., Rosenthal, G. G., & Przeworski, M. (2017). Repeated losses of PRDM9-directed recombination despite the conservation of PRDM9 across vertebrates. Elife, 6, e24133.
Battagin, M., Gorjanc, G., Faux, A. M., Johnston, S. E., & Hickey, J. M. (2016). Effect of manipulating recombination rates on response to selection in livestock breeding programs. Genetics Selection Evolution, 48(1), 1-12.


 , trait 2 only
, trait 2 only  , or both traits
, or both traits  , and are the defaults good?
, and are the defaults good?  ) … This means that you can look at single trait association data, and get an idea of the number of marginal associations, possibly dependent on allele frequency. The estimates from a gene expression dataset and a genome-wide association catalog work out to a prior around
) … This means that you can look at single trait association data, and get an idea of the number of marginal associations, possibly dependent on allele frequency. The estimates from a gene expression dataset and a genome-wide association catalog work out to a prior around  , which is the coloc default. So far so good.
, which is the coloc default. So far so good.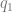 and
and 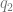 . But not all of the genome is functional. If you could straightforwardly define a functional proportion, you could just divide by it.
. But not all of the genome is functional. If you could straightforwardly define a functional proportion, you could just divide by it.