The European Society for Evolutionary Biology meeting this year took place August 20–25 in Groningen, Netherlands. As usual, the meeting was great, with lots of good talks and posters. I was also happy to meet colleagues, including people from Linköping who I’ve missed a lot since moving.
Here are some of my subjective highlights:
There were several interesting talks in the recombination symposium, spanning from theory to molecular biology and from within-population variation to phylogenetic distances. For example: Irene Tiemann-Boege talked about recombination hotspot evolution from the molecular perspective with mutation bias and GC-biased gene conversion (Arbeithuber & al 2015), while Franciso Úbeda de Torres presented a population genetic model model of recombination hotspots. I would need to pore over the paper to understand what was going on and if the model solves the hotspot paradox (as the title said), and how it is different from his previous model (Úbeda & Wilkins 2011).
There were also talks about young sex chromosomes. Alison Wright talked about recombination suppression on the evolving guppy sex chromosomes (Wright & al 2017), and Bengt Hansson about the autosome–sex chromosome fusion in Sylvioidea birds (Pala & al 2012).
Piter Bijma gave two (!) talks on social genetic effects. That is when your trait value depends not just on your genotype, but on the genotype on others around you, a situation that is probably not at all uncommon. After all, animals often live in groups, and plants have to stay put where they are. One can model this, which leads to a slightly whacky quantitative genetics where heritable variance can be greater than the trait variance, and where the individual and social effects can cancel each other out and prevent response to selection.
I first heard about this at ICQG in Edinburgh a few years ago (if memory serves, it was Bruce Walsh presenting Bijma’s slides?), but have only made a couple of fairly idle and unsuccessful attempts to understand it since. I got the feeling that social genetic effects should have some bearing on debates about kin selection versus multilevel selection, but I’m not sure how it all fits together. It is nice that it comes with a way to estimate effects (given that we know which individuals are in groups together and their relatedness), and there are some compelling case studies (Wade & al 2010). On the other hand, separating social genetic effects from other social effects must be tricky; for example, early social environment effects can look like indirect genetic effects (Canario, Lundeheim & Bijma 2017).
Philipp Gienapp talked about using realised relatedness (i.e. genomic relationships a.k.a. throw all the markers into the model and let partial pooling sort them out) to estimate quantitative genetic parameters in the wild. There is a lot of relevant information in the animal breeding and human genetics literature, but applying these things in the wild comes with challenges that deserves some new research to sort things out. Evolutionary genetics, similar to human genetics, is more interested in parameter estimation than prediction of phenotypes or breeding values. On the other hand, human genetics methods often work on GWAS summary statistics. In this way, evolutionary genetics is probably more similar to breeding. Also, the relatedness structure of the the populations may matter. Evolution happens in all kinds of populations, large and small, structured and well-mixed. Therefore, evolutionary geneticists may work with populations that are different from those in breeding and human genetics.
For example, someone asked about estimating genetic correlations with genomic relationships. There are certainly animal breeding and human genetics papers about realised relatedness and genetic correlation (Jia & Jannik 2012, Visscher & al 2014 etc), because of course, breeders need to deal a lot with correlated traits and human geneticists really like finding genetic correlations between different GWAS traits.
Speaking of population structure, Fst scans are still all the rage. There was a lot of discussion about trying to find regions of the genome that stand out as more differentiated in closely related populations (”genomic islands of speciation/divergence/differentiation”), and as less differentiated in mostly separated populations (introgression, possibly adaptive). But it’s not just Fst outliers. It’s encouraging to see different kinds of quantitative and population genomic methods applied in the same systems. On the hybrid and introgression side of things, Leslie Turner (Turner & Harr 2014) and Jun Kitano (Ravinet & al 2017) gave interesting talks on mice and sticklebacks, respectively. Danièle Filiaut showed an super impressive integrative GWAS and selection mapping study of local adaptation in Swedish Arabidopsis thaliana (Kedaffrec & al 2016).
Susan Johnston spoke about recombination mapping in Soay sheep and Rum deer (Johnston & al 2016, 2017). Given how few large long term genetic studies like this there are, it’s marvelous to be see the same kind of analysis in two parallel systems. Jason Munshi-South gave what seemed like a fascinating talk about rodent evolution in New York City (Harris & Munshi-South 2017). Unfortunately, too many other people thought so too, and I mostly failed to eavesdrop form the corridor.
Finally, Nina Wedell gave a wonderful presidential address about Evolution in the 21th century. ”Because I can. I’m the president now.” Yes!
The talk was about threats to evolutionary biology, examples of it’s usefulness and a series of calls to action. I liked the part about celebrating science much more than the common call to explain science to people. You know, like you hear at seminars and the march for science: We need to ”get out there” (where?) and ”explain what we’re doing” (to whom?). Because if it is true that science and scientists are being questioned, then scientists should speak in a way that works even if they’re not starting by default from a position of authority. Scientists need not just explain the science, but justify why the science is worth listening to in the first place.
”As your current president, I encourage you to celebrate evolution!”
I think this is precisely right, and it made me so happy. Of course, it leaves questions like ”What does that mean?”, ”How do we do it?”, but as a two word slogan, I think it is perfect.
Celebration aligns with sound rhetorical strategy in two ways. First, explanation is fine when someone asks for it, or is otherwise already disposed to listen to an explanation. But otherwise, it is more important to awaken interest and a positive state of mind before laying out the facts. (I can’t claim to be any kind of rhetorics expert. But see Rhetoric: for Herennius, Book I, V-VII for ancient wisdom on the topic.) By the way, I’m sure this is what people who are good at science communication actually do. Second, celebration means concentrating on the excitement and wonder, and the good things science can do. In that way, it prevents the trap of listing all the bad things that will happen if Trumpists, creationists and anti-vaccine activists get their way.
Nina Wedell also gave examples of the usefulness of evolution: biomimicry, directed evolution of enzymes, the power of evolutionary algorithms, plant and animal breeding, and prevention of resistance to herbicides and antibiotics. These are all good, worthy things, but also quite a limited subset of evolutionary biology? Maybe this idea is that evolutionary biology should be a basic science supporting applications like these. In line with that, she brought up how serendipitous useful things can come from studying strange diverse organisms and figuring out how they do things. The example in talk was the CRISPR–Cas system. Similar stories apply to a other proteins used as biomedical and biotechnology tools, such as Taq polymerase and Green fluorescent protein.
I have to question a remark about reproducibility, though. The list of threats included ”critique of the scientific method” and concerns over reproducibility, as if this was something that came from outside of science. I may have misunderstood. It was a very brief comment. But if problems with reproducibility are a threat to science, and I think they can be, then it’s not just a problem of image but a problem with how scientists perform, analyse, and report their science.
Evolutionary biology hasn’t been in the reproducibility crisis news the same way as psychology or behavioural genetics, but I don’t know if that is because of better quality, or just that no one has looked that carefully for the problems. There are certainly contradictory results here too, and the same overly flexible data analysis and selective reporting practices that cause problems elsewhere must be common in evolution too. I can think of some reasons why evolutionary biology may be better off. Parts of the field default to analysing data with multilevel or mixed models. Mixed models are not perfect, but they help with some multiple testing problems by fitting and partially pooling a lot of coefficients in the same model. Also, studies that use classical model organisms may be able to get a lot of replication, low variance, and large sample sizes in a way that is impossible for example with human experiments.
So I don’t know if there is a desperate need for large initiatives for replication of key results, preregistration of studies, and improvement of data analysis practice in evolution; there may or there may not. But wouldn’t it still be wonderful if we had them?
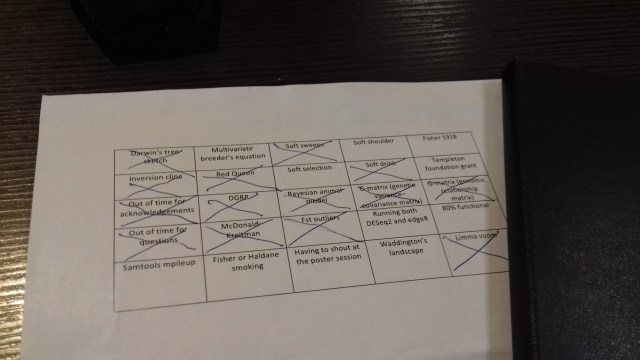
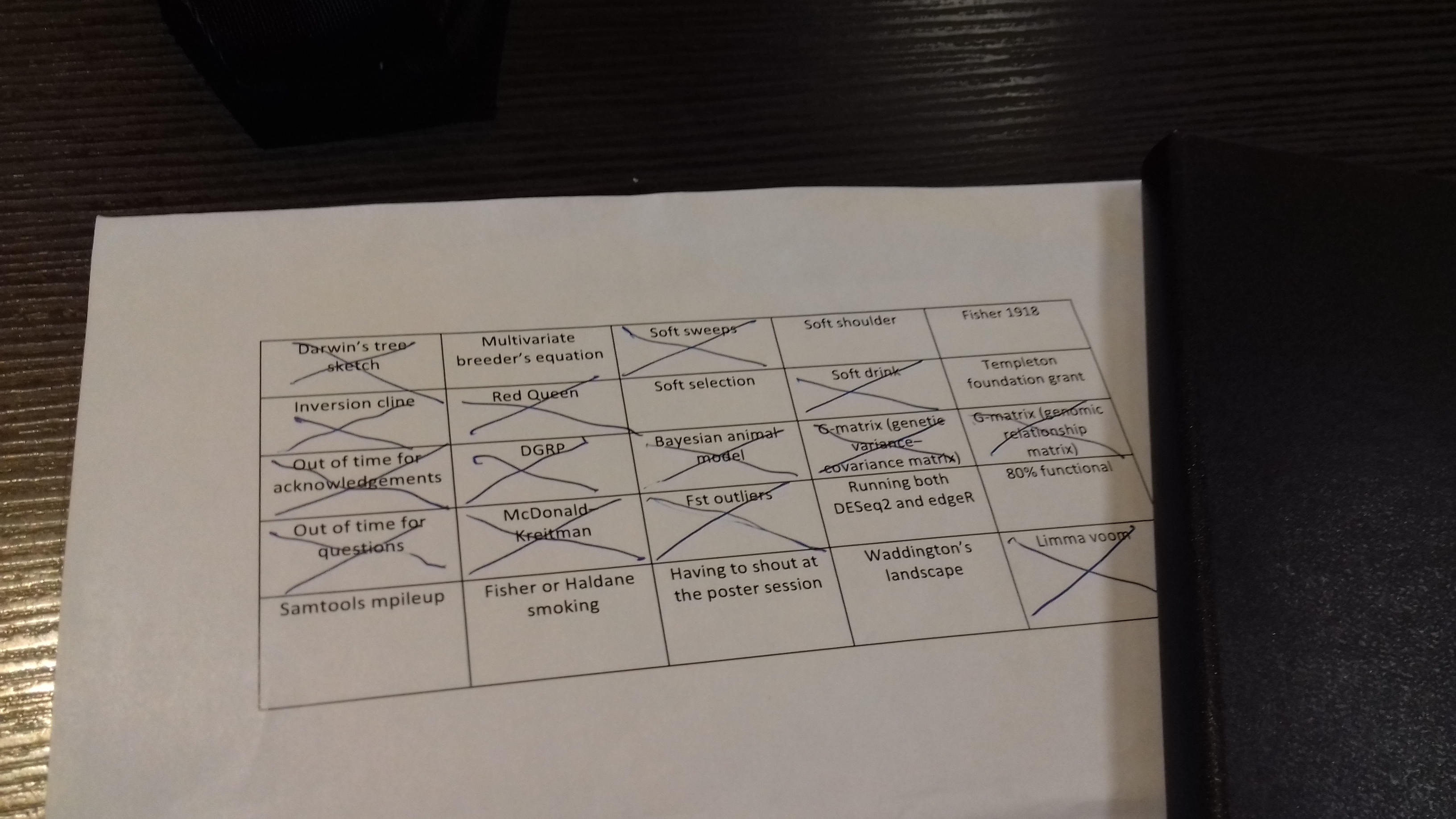
Bingo! I don’t have a ton of photos from Groningen, but here is my conference bingo card. Note what conspicuously isn’t filled in: the poster sessions took place in nice big room, and were not that loud. In retrospect, I probably didn’t go to enough of the non-genetic inheritance talks, and I should’ve put Fisher 1930 instead of 1918.

 and the ghost goes away. I would have been more interested in an attack on sophisticated adaptationism. How about the organismal level? Do ratchet-like neutral processes bias or direct the evolution of form and behaviour of say animals and plants?
and the ghost goes away. I would have been more interested in an attack on sophisticated adaptationism. How about the organismal level? Do ratchet-like neutral processes bias or direct the evolution of form and behaviour of say animals and plants?