I recently read this essay by Adrian Bird on ”The Selfishness of Law-Abiding Genes”. That is a colourful title in itself, but it doesn’t stop there; this is an extremely metaphor-rich piece. In terms of the theoretical content, there is not much new under the sun. Properties of the organism like complexity, redundancy, and all those exquisite networks of developmental gene regulation may be the result of non-adaptive processes, like constructive neutral evolution and intragenomic conflict. As the title suggests, Bird argues that this kind of thinking is generally accepted about things like transposable elements (”selfish DNA”), but that the same logic applies to regular ”law-abiding” genes. They may also be driven by other evolutionary forces than a net fitness gain at the organismal level.
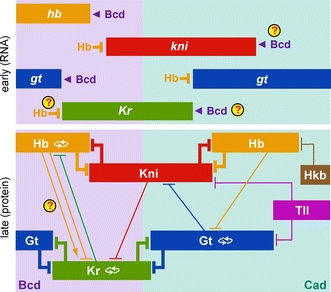
He gives a couple of possible examples: toxin–antitoxin gene pairs, RNA editing and MeCP2 (that’s probably Bird’s favourite protein that he has done a lot of work on). He gives this possible description of MeCP2 evolution:
Loss of MeCP2 via mutation in humans leads to serious defects in the brain, which might suggest that MeCP2 is a fundamental regulator of nervous system development. Evolutionary considerations question this view, however, as most animals have nervous systems, but only vertebrates, which account for a small proportion of the animal kingdom, have MeCP2. This protein therefore appears to be a late arrival in evolutionary terms, rather than being a core ancestral component of brain assembly. A conventional view of MeCP2 function is that by exerting global transcriptional restraint it tunes gene expression in neurons to optimize their identity, but it is also possible to devise a scenario based on self-interest. Initially, the argument goes, MeCP2 was present at low levels, as it is in non-neuronal tissues, and therefore played little or no role in creating an optimal nervous system. Because DNA methylation is sparse in the great majority of the genome, sporadic mutations that led to mildly increased MeCP2 expression would have had a minimal dampening effect on transcription that may initially have been selectively neutral. If not eliminated by drift, further chance increases might have followed, with neuronal development incrementally adjusting to each minor hike in MeCP2-mediated repression through compensatory mutations in other genes. Mechanisms that lead to ‘constructive neutral evolution’ of this kind have been proposed. Gradually, brain development would accommodate the encroachment of MeCP2 until it became an essential feature. So, in response to the question ‘why do brains need MeCP2?’, the answer under this speculative scenario would be: ‘they do not; MeCP2 has made itself indispensable by stealth’.
I think this is a great passage, and it can be read both as a metaphorical reinterpretation, and as substantive hypothesis. The empirical question ”Did MeCP2 offer an important innovation to vertebrate brains as it arose?”, is a bit hard to answer with data, though. On the other hand, if we just consider the metaphor, can’t you say the same about every functional protein? Sure, it’s nice to think of p53 as the Guardian of the Genome, but can’t it also be viewed as a gangster extracting protection money from the organism? ”Replicate me, or you might get cancer later …”
The piece argues for a gene-centric view, that thinks of molecules and the evolutionary pressures they face. This doesn’t seem so be the fashionable view (sorry, extended synthesists!) but Bird argues that it would be healthy for molecular cell biologists to think more about the alternative, non-adaptive, bottom-up perspective. I don’t think the point is to advocate that way of thinking to the exclusion of the all other. To me, the piece reads more like an invitation to use a broader set of metaphors and verbal models to aid hypothesis generation.
There are too may good quotes in this essay, so I’ll just quote one more from the end, where we’ve jumped from the idea of selfish law-abiding genes, over ”genome ecology” — not in the sense of using genomics in ecology, but in the sense of thinking of the genome as some kind of population of agents with different niches and interactions, I guess — to ”Genetics Meets Sociology?”
Biologists often invoke parallels between molecular processes of life and computer logic, but a gene-centered approach suggests that economics or social science may be a more appropriate model …
I feel like there is a circle of reinforcing metaphors here. Sometimes when we have to explain how something came to be, for example a document, a piece of computer code or a the we do things in an organisation, we say ”it grew organically” or ”it evolved”. Sometimes we talk about the genome as a computer program, and sometimes we talk about our messy computer program code as an organism. Like viruses are just like computer viruses, only biological.
Literature
Bird, Adrian. ”The Selfishness of Law-Abiding Genes.” Trends in Genetics 36.1 (2020): 8-13.