The blog has moved, and can now be found at: http://martinjohnsson.se/blog/
Old posts will remain here.
The blog has moved, and can now be found at: http://martinjohnsson.se/blog/
Old posts will remain here.
In the last post about conceptions of data-driven science, I wrote about the idea that reality is so complicated that we might need computers to help us reason about it. To be fair, even simple relationships in small data are too much for our puny brains to handle intuitively. We need to do the calculations. Here is Guest & Martin’s Pizza Problem of scientific reasoning, as presented in a thread by @cretiredroy on Twitter:
I ordered a 9-inch Pizza.
After a while, the waiter brought two 5-inch pizzas and said, the 9-inch pizza was not available and he was giving me two 5-inches Pizzas instead, and that I am getting 1 inch more for free!
The area of a circle is pi times the square of the radius r. A number n of 5-inch pizzas is smaller than a 9-inch pizza if:
Which we can rearrange to:
@cretiredroy again:
I said that even if he gave three pizzas, I would still lose-out.
”How can you say you are giving me an extra inch for free?”
The owner was speechless.
He finally gave me 4 pizzas.Take Maths seriously!
Guest & Martin (2021) use this simple problem as their illustration for computational model building: two 5 inch pizzas for the same price as one 9 inch pizza is not a good deal, because the 9 inch pizza contains more food. As I said before, I don’t think in inches and Swedish pizzas usually come in only one size, but this is counterintuitive to many people who have intuitions about inches and pizzas.
(In Guest & Martin's paper, the numbers are actually 12 and 18 inches. We can generalise to any radiuses of big and small pizzas:
Plugging in 12 and 18 gives 2.25, meaning that three 12-inch pizzas would be needed to sweeten the deal.)
We assume that we already agree that the relevant quantity is the area of pizza that we get. If we were out to optimise the amount of crust maybe the circumference, which is 2 times pi times the radius r, would be more relevant. In that case:


And the two smaller pizzas are a better deal with respect to crust circumference.
The risk of inconsistencies in our scientific understanding because we cannot intuitively grasp the implications of our models is what Guest & Martin call "The pizza problem". They believe that it can be ameliorated by computational modelling. If we have to make the calculations, we will notice and have to deal with assumptions we would otherwise not think about.
This is a different benefit of doing the calculations than the intuition building I wrote about in my post about the Monty Hall problem:
The outcome of the simulation is less important than the feeling that came over me as I was running it, though. As I was taking on the role of the host and preparing to take away one of the losing options, it started feeling self-evident that the important thing is whether the first choice is right. If the first choice is right, holding is the right strategy. If the first choice is wrong, switching is the right option. And the first choice, clearly, is only right 1/3 of the time.
I don’t think anyone is saying that it is impossible to intuitively grasp the case of the pizza deals. If you are used to relationships with squares, you might do it as quickly as @cretiredroy in the Twitter thread. However, because even simple power law relationships and probability experiments are tricky for us to reason about when we are naive to them, that should give us pause about our ability to reason verbally about new causal patterns. We might need some help.
Guest & Martin use the pizza example to illustrate their model of science as a hierarchy of frameworks, theories, specifications, implementations, hypotheses and data. The "framework" contains concepts of pizza, food and order, leading to the idea to maximize the amount of food per order. Our decision to care about amount of pizza and not about crust circumference belongs here. They describe two competing "theories": one that we need to maximise surface area and one that we need to maximise number of pizzas. The specification is the model of the area of a circle, and the implementation is plugging the numbers into it.
They argue that the "theory", "specification" and "implementation" steps are often omitted, and that doing so (prematurely jumping on to data collection), leads to inconsistencies that could have been avoided by being explicit about these steps. That is, in the pizza case, one should first write down the equations for the surface area of pizzas and compute the results before ordering the two deals and weighing the pizzas.
Just writing down the equations is not enough. In their talk on the topic for Glasgow ReproducibiliTea, Guest & Martin (2020) emphasise that the pizza problem is an issue even when we already have a formal model that describes the problem well — as in the pizza case, everyone agrees on the maths of the area of a circle — but that does not prevent the result from being counterintuitive. The formal model does not actually help resolve the confusion until it is used to compute the area of the two pizza orders.



I wanted to illustrate this post with a photo of a pizza from the chippy in Penicuik, where you get a complimentary box of chips with the larger size pizza, but I couldn’t find it. Instead, here are some pizzas from #GeneticistsTweetingAboutPizza.
References
@cretiredroy Twitter thread https://twitter.com/cretiredroy/status/1542351846903529472
Guest, O., & Martin, A. E. (2021). How computational modeling can force theory building in psychological science. Perspectives on Psychological Science, 16(4), 789-802.
Guest O & Martin (2020). How computational modeling can force theory building in psychological science (Glasgow ReproTea). Recording. https://www.youtube.com/watch?v=_WV7EFvFAB8
This list of gradually more radical conceptions represent my attempt at understanding the term ”data-driven science”. What does it mean?
This is the non-answer that I expect from researchers who take ”data-driven” to mean ”empirical science as usual”, potentially distinguishing it from ”theory”, which they may dislike.
This is the answer I expect from the ”milk recordings are Big Data” crew, i.e., researchers who are used to using large datasets, and don’t see anything truly new in the ”Big Data” movement (but might like to take part in the hype with their own research).
For example, Leonelli (2014) writes about developments in model organism databases and their relation to ”Big Data”. She argues that working with large datasets was nothing new for this community but that the Big Data discourse brought attention to the importance of data and data management:
There is strong continuity with practices of large data collection and assemblage conducted since the early modern period; and the core methods and epistemic problems of biological research, including exploratory experimentation, sampling and the search for causal mechanisms, remain crucial parts of inquiry in this area of science – particularly given the challenges encountered in developing and applying curatorial standards for data other than the high-throughput results of ‘omics’ approaches. Nevertheless, the novel recognition of the relevance of data as a research output, and the use of technologies that greatly facilitate their dissemination and re-use, provide an opportunity for all areas in biology to reinvent the exchange of scientific results and create new forms of inference and collaboration.
This answer is the notion that data-driven research is about making use of the data that is already out there as much as possible, putting it in new research contexts and possibly aggregating data from many sources.
This is the emphasis of the SciLife & Wallenberg program for ”data-driven life science”:
Experiments generate data, which can be analyzed to address specific hypotheses. The data can also be used and combined with other data, into larger and more complex sets of information, and generate new discoveries and new scientific models. Which, in their turn, can be addressed with new experiments.
Researchers in life science often collect large amounts of data to answer their research questions. Often, the same data can be used to answer other research questions, posed by other research groups. Perhaps many, many other questions, and many, many other research groups. This means that data analysis, data management and data sharing is central to each step of the research process …
In my opinion, taking this stance does not necessarily imply any ban on modelling, hypothesising or theorising (which is the next level). It’s a methodological emphasis on data, not a commitment to a particular philosophy of science.
This answer understands ”data-driven science” as a philosophy of science position that prioritises data exploration over modelling, hypothesising and theorising. Leonelli argues that this is one of the key features of data driven research (2012).
Extreme examples include Chris Anderson’s view that ”correlation is enough” and that large datasets have made theories and searches for causes obsolete. Anderson wrote in Wire 2008 about how ”petabyte-scale” data is making theory, hypotheses, models, and the search for causes obsolete.
The new availability of huge amounts of data, along with the statistical tools to crunch these numbers, offers a whole new way of understanding the world. Correlation supersedes causation, and science can advance even without coherent models, unified theories, or really any mechanistic explanation at all.
But faced with massive data, this approach to science — hypothesize, model, test — is becoming obsolete. Consider physics: Newtonian models were crude approximations of the truth (wrong at the atomic level, but still useful). A hundred years ago, statistically based quantum mechanics offered a better picture — but quantum mechanics is yet another model, and as such it, too, is flawed, no doubt a caricature of a more complex underlying reality. The reason physics has drifted into theoretical speculation about n-dimensional grand unified models over the past few decades (the ”beautiful story” phase of a discipline starved of data) is that we don’t know how to run the experiments that would falsify the hypotheses — the energies are too high, the accelerators too expensive, and so on.
/…/
There is now a better way. Petabytes allow us to say: ”Correlation is enough.” We can stop looking for models. We can analyze the data without hypotheses about what it might show. We can throw the numbers into the biggest computing clusters the world has ever seen and let statistical algorithms find patterns where science cannot.
I believe that this extreme position is incoherent. We can’t infer directly from data without a theory or model. The options are to do it with or without acknowledging and analysing the models we use. But there are less extreme versions that make more sense.
For example, van Helden (2013) argued that omic studies imply more broad and generic hypotheses, about what kinds of mechanisms and patterns one might expect to find in a large dataset, rather than hypothesising the identity of the genes, pathways or risk factors themselves.
Equipped with such huge data sets, we can perform data mining in an objective way. For some purists, this approach to data acquisition is anathema, as it is not ‘hypothesis-driven’. However, I submit that it is. In this case, the original hypothesis is broad or generic–we generate data, assess it and probably find something useful for elucidating our research problem. /…/ The hypothesis is that one will design an algorithm and find a pattern, which allows us to distinguish between cases and controls.
Anderson might be right that the world as revealed by data is too complicated for our puny brains to write down equations about; maybe machines can help. That brings us to the next level.
This answer is the attitude that complex relationships in large data are too much for the human mind, and traditional theoretical models, to handle. Instead, we should use machine learning models to find patterns and make predictions using automated inference processes that are generally too intricate for humans to interpret intuitively. This is also one of Leonelli’s key features of data-driven research (2012).
Again, Anderson’s ”End of Theory” is an extreme example of this view, when he argues that the complex interactions between genes, epigenetics and environment make theoretical models of genetics useless.
Now biology is heading in the same direction. The models we were taught in school about ”dominant” and ”recessive” genes steering a strictly Mendelian process have turned out to be an even greater simplification of reality than Newton’s laws. The discovery of gene-protein interactions and other aspects of epigenetics has challenged the view of DNA as destiny and even introduced evidence that environment can influence inheritable traits, something once considered a genetic impossibility.
In short, the more we learn about biology, the further we find ourselves from a model that can explain it.
Anderson overstates how helpless genetic models are against complexity, but there is a point here, for example with regard to animal breeding. Part of what made genomics useful in modern animal breeding was giving up on the piecemeal identification of individual markers of large genetic effect, and instead throwing whole-genome information into a large statistical model. We estimate them all, without regard for their individual effect, without looking for genetic causes (”genetics without genes” as Lowe & Bruce (2019) put it), and turn them into a ranking of animals for selection decisions. However, this is level 3 stuff, because there is a human-graspable quantitative genetic theory underlying the whole machinery, with marker effect, breeding values, and equations that can be interpreted. A more radical vision of machine learning-based breeding would take away that too, no notion of genetic relationship between individuals, genetic correlation between traits, or separating genotype from phenotype. Will this work? I don’t know, but there are tendencies like this in the field, for example when researchers try to predict useful traits from high-dimensional omics or sensor data.
Apart from those pro-machine learning, anti-theory stances, you could have a pro-machine pro-theory stance. There several fields of research that try to use automated reasoning to help do theory. One interesting subfield of research that I only learned about recently is research around ”symbolic regression” and ”machine scientists” where you try to find mathematical expressions that fit data but allowing for a large variety of different expressions. It’s like a form of model selection where you allow a lot of flexibility about what kind of model you consider.
There is a whole field of this, but here is an example I read recently: Guimerà et al. (2020) used a Bayesian method, where they explore different expression with Markov Chain Monte Carlo, putting a prior on the mathematical expression based on equations from Wikipedia. This seems like something that could be part of a toolbox of a data-driven scientist who still won’t give up on the idea that we can understand what’s going on, even if we might need a help from a computer.
Anderson C., The End of Theory: The Data Deluge Makes the Scientific Method Obsolete. Wired.
van Helden P., 2013 Data-driven hypotheses. EMBO reports 14: 104–104.
Leonelli S., 2012 Introduction: Making sense of data-driven research in the biological and biomedical sciences. Studies in History and Philosophy of Science Part C: Studies in History and Philosophy of Biological and Biomedical Sciences 43: 1–3.
Leonelli S., 2014 What difference does quantity make? On the epistemology of Big Data in biology. Big Data & Society 1: 2053951714534395.
Lowe, James WE, and Ann Bruce., 2019 Genetics without genes? The centrality of genetic markers in livestock genetics and genomics. History and Philosophy of the Life Sciences 41.4
Guimerà, R., Reichardt, I., Aguilar-Mogas, A., Massucci, F. A., Miranda, M., Pallarès, J., & Sales-Pardo, M., 2020. A Bayesian machine scientist to aid in the solution of challenging scientific problems. Science advances, 6(5), eaav6971.
A few months ago I saw this nice figure from Amy Williams of the number of DNA segments that are expected to be shared between relatives. I thought it would be fun to simulate segment sharing with AlphaSimR.
Because DNA comes in chromosomes that don’t break up and recombine that much, the shared DNA between relatives tends to come in long chunks — segments that are identical by descent. The distribution of segment lengths can sometimes be used to tell apart relationships that would otherwise give the same average (e.g., Yengo et al. 2019, Qiao et al. 2021).
But let’s not do anything sophisticated. Instead, we take three very simple pedigrees — anyone who’s taken introductory genetics will recognize these ones — and look at relationships between full-sibs, half-sibs and cousins. We’ll also look at the inbred offspring of matings between full-sibs, half-sibs and cousins to see that the proportion that they share between their two copies of the genome lines up with the expected inbreeding.
There won’t be any direct comparison to the values that Williams’ simulation, because it simulated more distant relationships than this, starting with cousins and then moving further away. This is probably more interesting, especially for human genealogical genetics.
The code is on GitHub if you wants to follow along.
Here are the three pedigrees, drawn with the kinship2 package:
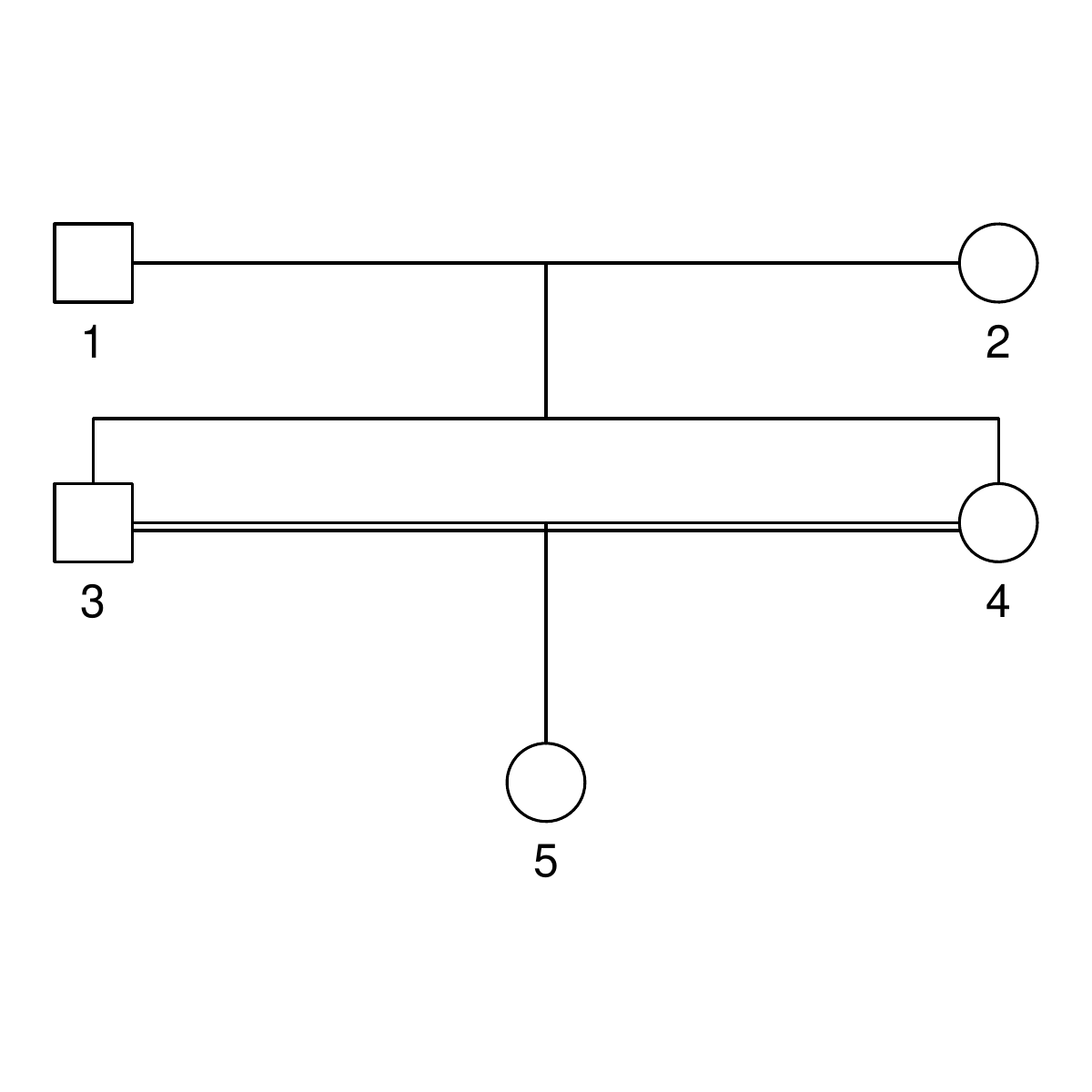
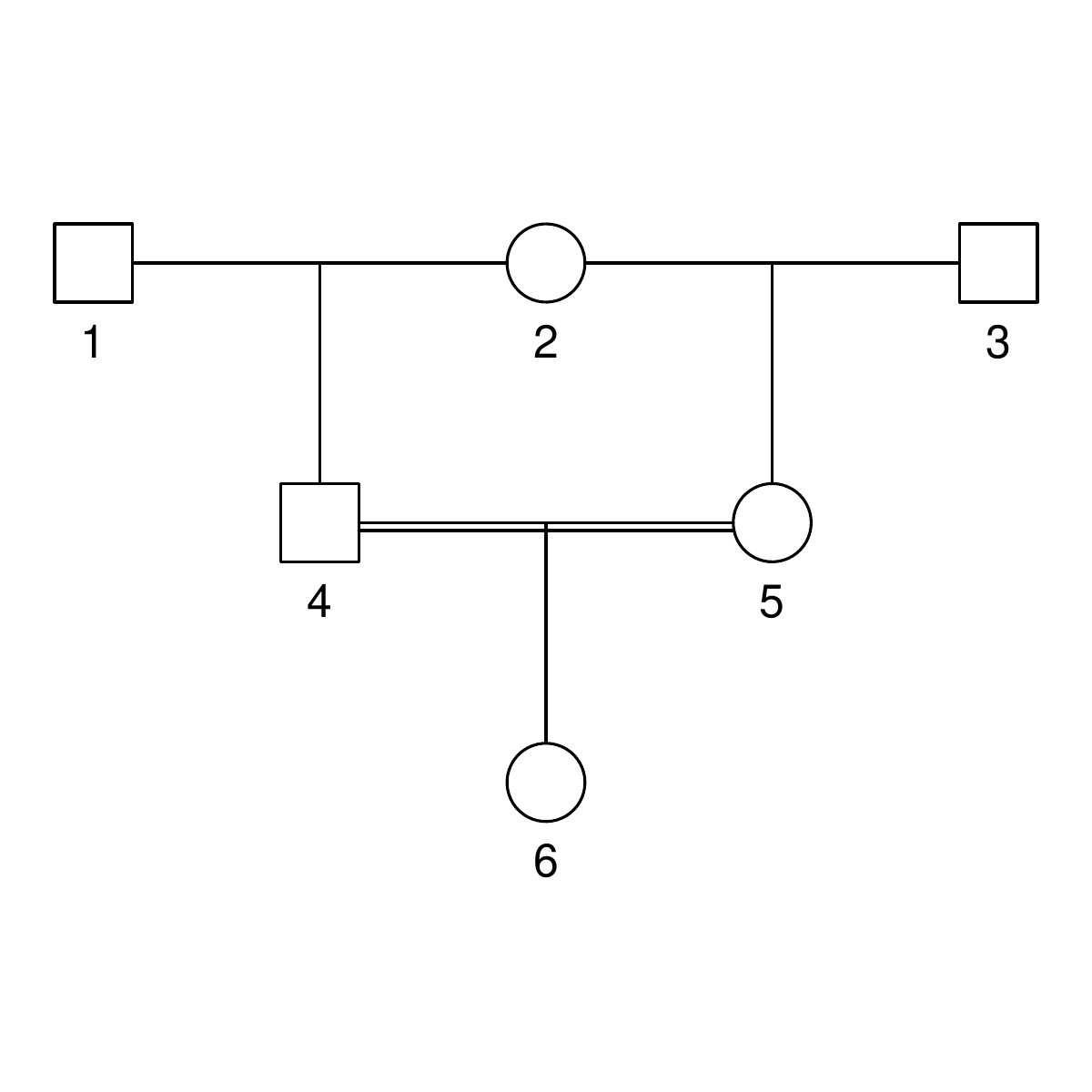
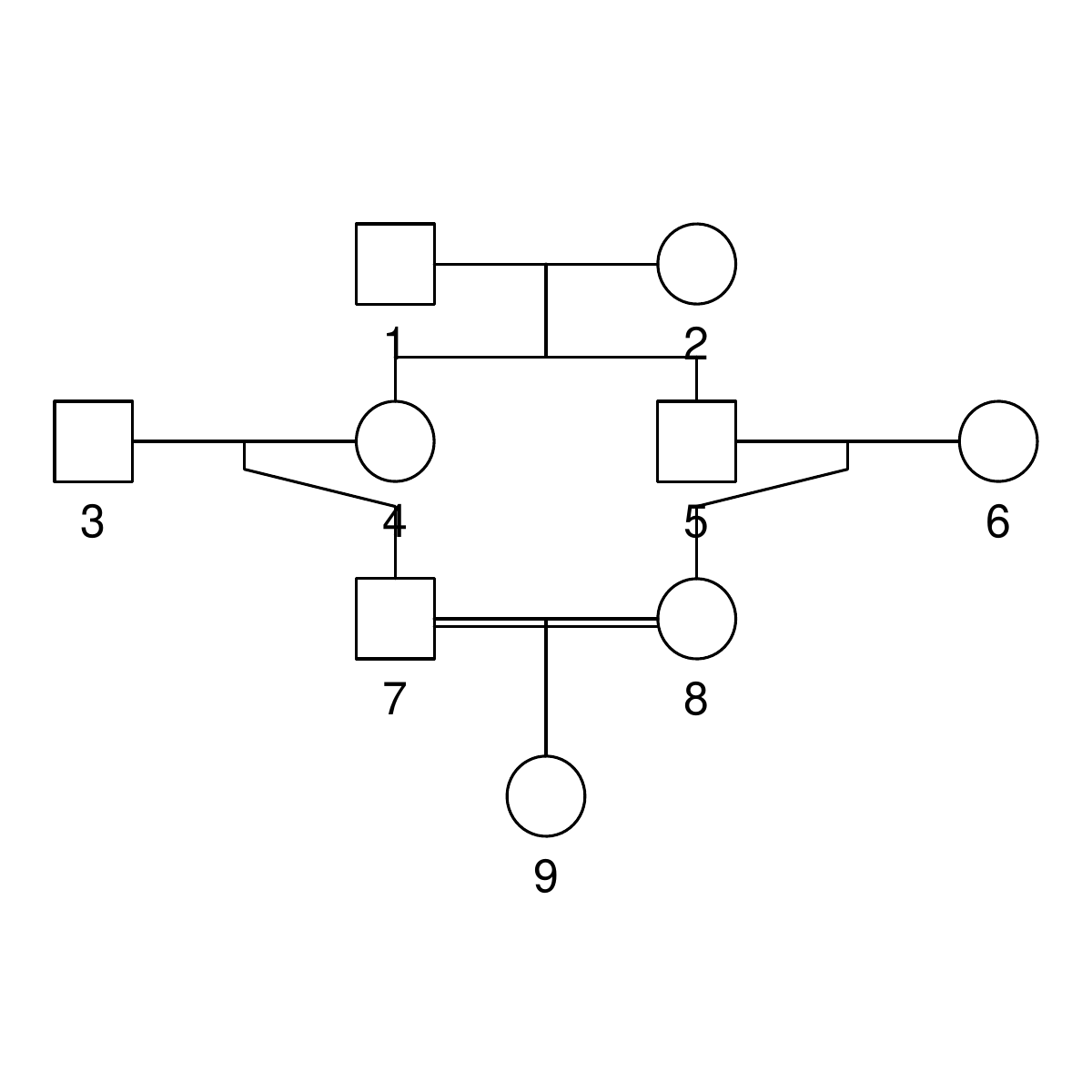
A pedigree, here, is really a table of individuals, where each column tells us their identifier, their parents, and optionally their sex, like this:
id, mother, father, sex 1, NA, NA, M 2, NA, NA, F 3, NA, NA, M 4, 2, 1, F 5, 2, 1, M 6, NA, NA, F 7, 4, 3, M 8, 6, 5, F 9, 8, 7, F
We can use GeneticsPed to check the relatedness and inbreeding if we don’t trust that I’ve entered the pedigrees right.
library(GeneticsPed)
library(purrr)
library(readr)
ped_fullsib <- read_csv("pedigrees/inbreeding_fullsib.txt")
ped_halfsib <- read_csv("pedigrees/inbreeding_halfsib.txt")
ped_cousin <- read_csv("pedigrees/inbreeding_cousin.txt")
inbreeding_ped <- function(ped) {
inbreeding(Pedigree(ped))
}
print(map(list(ped_fullsib, ped_halfsib, ped_cousin), inbreeding_ped))
[[1]]
1 2 3 4 5
0.00 0.00 0.00 0.00 0.25
[[2]]
1 2 3 4 5 6
0.000 0.000 0.000 0.000 0.000 0.125
[[3]]
1 2 3 4 5 6 7 8 9
0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0625
We need some functions to compare haplotypes and individuals:
library(AlphaSimR)
library(dplyr)
library(purrr)
library(tibble)
## Find shared segments between two haplotypes expressed as vectors
## map is a vector of marker positions
compare_haplotypes <- function(h1, h2, map) {
sharing <- h1 == h2
runs <- rle(sharing)
end <- cumsum(runs$lengths)
start <- c(1, end[-length(end)] + 1)
segments <- tibble(start = start,
end = end,
start_pos = map[start],
end_pos = map[end],
segment_length = end_pos - start_pos,
value = runs$values)
segments[segments$value,]
}
We will have haplotypes of the variants that go together on a chromosome, and we want to find segments that are shared between them. We do this with a logical vector that tests each variant for equality, and then use the rle to turn this into run-length encoding. We extract the start and end position of the runs and then keep only the runs of equality.
Building on that function, we want to find the shared segments on a chromosome between two individuals. That is, we make all the pairwise comparisons between the haplotypes they carry and combine them.
## Find shared segments between two individuals (expressed as
## matrices of haplotypes) for one chromosome
compare_individuals_chr <- function(ind1, ind2, map) {
h1_1 <- as.vector(ind1[1,])
h1_2 <- as.vector(ind1[2,])
h2_1 <- as.vector(ind2[1,])
h2_2 <- as.vector(ind2[2,])
sharing1 <- compare_haplotypes(h1_1, h2_1, map)
sharing2 <- compare_haplotypes(h1_1, h2_2, map)
sharing3 <- compare_haplotypes(h1_2, h2_1, map)
sharing4 <- compare_haplotypes(h1_2, h2_2, map)
bind_rows(sharing1, sharing2, sharing3, sharing4)
}
Finally, we use that function to compare individuals along all the chromosomes.
This function takes in a population and simulation parameter object from AlphaSimR, and two target individuals to be compared.
We use AlphaSimR‘s pullIbdHaplo function to extract tracked founder haplotypes (see below) and then loop over chromosomes to apply the above comparison functions.
## Find shared segments between two target individuals in a
## population
compare_individuals <- function(pop,
target_individuals,
simparam) {
n_chr <- simparam$nChr
ind1_ix <- paste(target_individuals[1], c("_1", "_2"), sep = "")
ind2_ix <- paste(target_individuals[2], c("_1", "_2"), sep = "")
ibd <- pullIbdHaplo(pop,
simParam = simparam)
map <- simparam$genMap
loci_per_chr <- map_dbl(map, length)
chr_ends <- cumsum(loci_per_chr)
chr_starts <- c(1, chr_ends[-n_chr] + 1)
results <- vector(mode = "list",
length = n_chr)
for (chr_ix in 1:n_chr) {
ind1 <- ibd[ind1_ix, chr_starts[chr_ix]:chr_ends[chr_ix]]
ind2 <- ibd[ind2_ix, chr_starts[chr_ix]:chr_ends[chr_ix]]
results[[chr_ix]] <- compare_individuals_chr(ind1, ind2, map[[chr_ix]])
results[[chr_ix]]$chr <- chr_ix
}
bind_rows(results)
}
(You might think it would be more elegant, when looping over chromosomes, to pull out the identity-by-descent data for each chromosome at a time. This won’t work on version 1.0.4 though, because of a problem with pullIbdHaplo which has been fixed in the development version.)
We use an analogous function to compare the haplotypes carried by one individual. See the details on GitHub if you’re interested.
We are ready to run our simulation: This code creates a few founder individuals that will initiate the pedigree, and sets up a basic simulation. The key simulation parameter is to set setTrackRec(TRUE) to turn on tracking of recombinations and founder haplotypes.
source("R/simulation_functions.R")
## Set up simulation
founders <- runMacs(nInd = 10,
nChr = 25)
simparam <- SimParam$new(founders)
simparam$setTrackRec(TRUE)
founderpop <- newPop(founders,
simParam = simparam)
To simulate a pedigree, we use pedigreeCross, a built-in function to simulate a given pedigree, and then apply our comparison functions to the resulting simulated population.
## Run the simulation for a pedigree one replicate
simulate_pedigree <- function(ped,
target_individuals,
focal_individual,
founderpop,
simparam) {
pop <- pedigreeCross(founderPop = founderpop,
id = ped$id,
mother = ped$mother,
father = ped$father,
simParam = simparam)
shared_parents <- compare_individuals(pop,
target_individuals,
simparam)
shared_inbred <- compare_self(pop,
focal_individual,
simparam)
list(population = pop,
shared_segments_parents = shared_parents,
shared_segments_self_inbred = shared_inbred)
}
First we can check how large proportion of the genome of our inbred individuals is shared between their two haplotypes, averaged over 100 replicates. That is, how much of the genome is homozygous identical by descent — what is their genomic inbreeding? It lines up with the expectation form pedigree: 0.25 for the half-sib pedigree, close to 0.125 for the full-sib pedigree and close to 0.0625 for the cousin pedigree. The proportion shared by the parents is, as it should, about double that.
case inbred_self_sharing parent_sharing
full-sib 0.25 (0.052) 0.5 (0.041)
half-sib 0.13 (0.038) 0.25 (0.029)
cousin 0.064 (0.027) 0.13 (0.022)
Table of the mean proportion of genome shared between the two genome copies in inbred individuals and between their parents. Standard deviations in parentheses.
This is a nice consistency check, but not really what we wanted. The point of explicitly simulating chromosomes and recombinations is to look at segments, not just total sharing.
With a little counting and summarisation, we can plot the distributions of segment lengths. The horizontal axis is the length of the segments expressed in centimorgan. The vertical axis is the number of shared
segments of this length or longer. Each line is a replicate.

If we look at the summaries (table below), full-sibs share on average 74 segments greater than 1 cM in length, half-sibs 37, and cousins 29.
In real data, short segments might be harder to detect, but because we’re using simulated fake data, we don’t have to worry about phasing errors or false positive sharing.
If we look only at long segments (> 20 cM), full-sibs share on average 46 segments, half-sibs 23, and cousins 13. (Also, similar to Williams’ simulations, none of the cousins simulated here had less than five long segments shared.)
case `1 cM` `10 cM` `20 cM` `30 cM` `40 cM` full-sib 74 (5.2) 60 (4.2) 46 (3.6) 34 (4) 24 (3.8) half-sib 37 (3.4) 30 (3.1) 23 (3.3) 17 (2.8) 13 (2.6) cousin 29 (3.8) 20 (3.3) 13 (3.2) 7.6 (2) 4.3 (1.8)
Table of the mean number of shared segments of different minimum length. Standard deviations in parentheses.
We an also look at the average length of the segments shared, and note that while full-sibs and half-sibs differ in the number of segments, and total segment length shared (above), the length of individual segments is about the same:
case mean_length_sd full-sib 0.33 (0.032) half-sib 0.34 (0.042) cousin 0.21 (0.03)
Table of the mean length shared segments. Standard deviations in parentheses.
Williams’ simulation, using the ped-sim tool, had a more detailed model of recombination in the human genome, with different interference parameters for each chromosome, sex-specific recombination and so on. In that way, it is much more realistic.
We’re not modelling any one genome in particular, but a very generic genome. Each chromosome is 100 cM long for example; one can imagine that a genome with many short chromosomes would give a different distribution. This can be changed, though; the chromosome size is the easiest, if we just pick a species.
Literature
Yengo, L., Wray, N. R., & Visscher, P. M. (2019). Extreme inbreeding in a European ancestry sample from the contemporary UK population. Nature communications, 10(1), 1-11.
Qiao, Y., Sannerud, J. G., Basu-Roy, S., Hayward, C., & Williams, A. L. (2021). Distinguishing pedigree relationships via multi-way identity by descent sharing and sex-specific genetic maps. The American Journal of Human Genetics, 108(1), 68-83.
Without making any predictions about what will happen with the pandemic or when, I suspect that we are never going back to the way things were in terms of on-campus education and meetings. A lot more of university life will be hybrid online. This isn’t because hybrid online is necessarily great; I suspect that in terms of meetings, a hybrid meeting with some folks in a room and others on videoconference is the worst of two worlds. But it might be the best option that is possible — when an in-person meeting is better than a hybrid meeting, but a hybrid meeting is better than no meeting.
People get sick, at any time, and the prosocial thing to do is to stay at home and not spread the infection more than necessary. I expect, and hope, that this norm will remain, and we will have less people sick in class and at work. Students who fall ill will, reasonably, expect to be able to participate from home when they do the right thing and stay home from class. Teachers who suddenly fall ill likely expect it too. After all, it is less painful, for everyone involved, than cancelling and rescheduling.
If we are reasonable and empathetic, we will accommodate. If I’m right that hybrid meetings are, in general, slightly worse than in person meetings, the meeting quality will on average be worse — but more people will be able to participate, and that is also valuable. So it might not feel like it, but taken together, the hybrid solution might be better. The exact balance would depend on how much worse or better different meeting forms are; we probably can’t put numbers on that.
The same argument applies to online scientific conferences. I can’t imagine that the online conference experience is as useful, inspiring and conducive to networking as an in-person conference. My personal impression is that online conferences and seminars are great for watching talks, but bad for meeting people. However, online conferences are accessible to more people — those who for some reason can’t travel, can’t afford it, can’t be away from home for that long.
We might not feel excited about it, but the hybrid online future is here.
Dear diary,
Time for the meta-post of the year! During 2021, the blog racked up 28 posts (counting this one), about the same pace as 2020. That’s okay.
As usual, let’s pick one post a month to represent the blogging year of 2021:
January: A model of polygenic adaptation in an infinite population. We look at some equations and make two animations following Jain & Stephan (2017).
February: The Fulsom Principle: Smart people will gladly ridicule others for breaking supposed rules that are in fact poorly justified.
March: Theory in genetics. As I’m using more modelling in my work, here is some inspiration from an essay by Brian Charlesworth. As I hadn’t read Guest & Martin (2021) at the time, the post only cites Robinaugh at al. (2020) and music theory youtuber Adam Neely, and that’s also quite good.
April: Researchers in ecology and evolution don’t use Platt’s strong inference, and that’s okay. This post is reacting to a paper that advocates explicit hypotheses in evolution and ecology, a paper I think qualifies as ”wrong, but wrong in an interesting and productive way” (can’t remember who said that).
Also, this post also got one of the coveted Friday Links mentions on Dynamic Ecology, before it shut down. End of an era, death of blogs etc.
May: Convincing myself about the Monty Hall problem Probably my favourite post of the year. It also shows the value of a physical model: I had what felt like the crucial moment that made the problem click as I was physically acting out Monty Hall choices with myself. Also, first shout-out to Guest & Martin (2021), a paper that will be cited again on the blog, I’m sure. I think the Monty Hall problem is a great example of their claim that a precise mathematical theory can’t defeat intuition unless you run the numbers and do the calculations.
June: no posts, distracted by work and pandemic, I guess.
July: Journal club of one: ”A unifying concept of animal breeding programmes”. Rather long post about a paper about a graphical method for displaying simulated breeding structures, and whether this qualifies as a formal specification or not.
August: ”Dangerous gene myths abound” , reacting to an article by Philip Ball. The breathless hype around genomics is often embarrassing, but criticisms of reductionism in genetics love attacking caricatures.
September: Belief in science, some meditations on how little it knows and how science ”works whether you believe in it or not”
October–November: extended blog vacation
December: Well, there are only two to choose from, but the other day I posted some notes about two methods that try to infer recent population history from linkage disequilibrium.
Outwith the blog, there were also some papers published. I’m really behind on giving them their own blog posts, but that might be rectified in time. The blog runs on its own schedule where papers remain ”recent” for several years.
What else is new? Astute readers may have noticed that my job description has changed from ”postdoc” to ”researcher”. Now, ”researcher” is a little bit of an ambiguous title because some institutions also use it for time limited positions — but no, dear reader, I am now permanently employed at the Department of Animal Breeding and Genetics, Swedish University of Agricultural Sciences where, among other things, I lead the ”Genome dynamics of livestock breeding” project financed by the research council Formas. This will be great fun, and also show on the blog in time, I’m sure.
In this post, we will look at running two programs that infer population history — understood as changes in linkage disequilibrium over time — from genotype data. The post will chronicle running them on some simulated data; it will be light on theory, and light on methods evaluation.
Linkage disequilibrium, i.e. correlation between alleles at different genetic variants, breaks down over time when alleles are shuffled by recombination. The efficiency of that process depends on the distance between the variants (because variants close to each other on the same chromosome will recombine less often) and the population size (because more individuals means more recombinations). Those relationships mean that the strength of linkage disequilibrium at a particular distance between variants is related to the population size at a particular time. (Roughly, and assuming a lot.) There are several methods that make use of the relationship between effective population size, recombination rate and linkage disequilibrium to estimate population history.
The two programs we’ll look at are SNeP and GONE. They both first calculate different statistics of pairwise linkage disequilibrium between markers. SNeP groups pairs of markers into groups based on the distance between them, and estimates the effective population size for each group and how many generations ago each group represents. GONE goes further: it uses a formula for the expected linkage disequilibrium from a sequence of effective population sizes and a genetic algorithm to find such a sequence that fits the observed linkage disequilibrium at different distances.
Paper about GONE: Santiago, E., Novo, I., Pardiñas, A. F., Saura, M., Wang, J., & Caballero, A. (2020). Recent demographic history inferred by high-resolution analysis of linkage disequilibrium. Molecular Biology and Evolution, 37(12), 3642-3653.
Paper about SNeP: Barbato, M., Orozco-terWengel, P., Tapio, M., & Bruford, M. W. (2015). SNeP: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Frontiers in genetics, 6, 109.
These methods are suited for estimating recent population history in single closed populations. There are other methods, e.g. the Pairwise Markovian coealescent and methods based on Approximate Bayesian Computation, that try to reach further back in time or deal with connected populations.
(Humorously, barring one capitalisation difference, GONE shares it’s name with an unrelated program related to effective population sizes, GONe … There are not enough linkage disequilibrium puns to go around, I guess.)
First, let us generate some fake data to run the programs on. We will use the Markovian coalescent simulator MaCS inside AlphaSimR. That is, we won’t really make use of any feature of AlphaSimR except that it’s a convenient way to run MaCS.
There is a GitHub repo if you want to follow along.
We simulate a constant population, a population that decreased in size relatively recently, a population that increased in size recently, and a population that decreased in longer ago. The latter should be outside of what these methods can comfortably estimate. Finally, let’s also include a population that has migration from an other (unseen) population. Again, that should be a case these methods struggle with.

Simulated true population histories. Note that the horizontal axis is generations ago, meaning that if you read left to right, it runs backwards in time. This is typical when showing population histories like this, but can be confusing. Also not the different scales on the horizontal axis.
library(AlphaSimR)
library(purrr)
library(tibble)
## Population histories
recent_decrease <- tibble(generations = c(1, 50, 100, 150),
Ne = c(1000, 1500, 2000, 3000))
recent_increase <- tibble(generations = c(1, 50, 100, 150),
Ne = c(3000, 2000, 1500, 1000))
ancient_decrease <- tibble(generations = recent_decrease$generations + 500,
Ne = recent_decrease$Ne)
We can feed these population histories (almost) directly into AlphaSimR’s runMacs2 function. The migration case is a little bit more work because we will to modify the command, but AlphaSimR still helps us. MaCS takes a command line written using the same arguments as the paradigmatic ms program. The runMacs2 function we used above generates the MaCS command line for us; we can ask it to just return the command for us to modify. The split argument tells us that we want two populations that split 100 generations ago.
runMacs2(nInd = 100,
Ne = recent_decrease$Ne[1],
histGen = recent_decrease$generations[-1],
histNe = recent_decrease$Ne[-1],
split = 100,
returnCommand = TRUE)
The resulting command looks like this:
"1e+08 -t 1e-04 -r 4e-05 -I 2 100 100 -eN 0.0125 1.5 -eN 0.025 2 -eN 0.0375 3 -ej 0.025001 2 1"
The first part is the number of basepairs on the chromosome, -t flag is for the population mutation rate 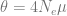
-r for the recombination rate (also multiplied by four times the effective population size). The -eN arguments change the population size, and the -ej argument is for splitting and joining populations.
We can check that these numbers make sense: The population mutation rate of 10-4 is the typical per nucleotide mutation rate of 2.5 × 10-8 multiplied by four times the final population size of 1000. The scaled recombination rate of 4 × 10-5 is typical per nucleotide recombination rate of 10-8 multiplied by the same.
The population size changes (-eN arguments) are of the format scaled time followed by a scaling of the final population size. Times are scaled by four times the final population size, again. This means that 0.0125 followed by 1.5 refers to that 4 × 0.0125 × 1000 = 50 generations ago population size was 1.5 times the final population size, namely 1500. And so on.
-I (I assume for ”island” as in the ”island model”) sets up the two populations, each with 100 individuals and a migration rate between them. We modify this by setting it to 200 for the first population (because we want diploid individuals, so we need double the number of chromosomes) and 0 for the other; that is, this ghost population will not be observed, only used to influence the first one by migration. Then we set the third parameter, that AlphaSimR has not used: the migration rate. Again, this is expressed as four times the final population size, so for a migration rate of 0.05 we put 200.
Now we can run all cases to generate samples of 100 individuals.
migration_command <- "1e+08 -t 1e-04 -r 4e-05 -I 2 200 0 200 -eN 0.0125 1.5 -eN 0.025 2 -eN 0.0375 3 -ej 0.025001 2 1"
pops <- list(pop_constant = runMacs2(nInd = 100,
nChr = 5,
histNe = NULL,
histGen = NULL,
Ne = 1000),
pop_recent = runMacs2(nInd = 100,
nChr = 5,
Ne = recent_decrease$Ne[1],
histGen = recent_decrease$generations[-1],
histNe = recent_decrease$Ne[-1]),
pop_increase = runMacs2(nInd = 100,
nChr = 5,
Ne = recent_increase$Ne[1],
histGen = recent_increase$generations[-1],
histNe = recent_increase$Ne[-1]),
pop_ancient = runMacs2(nInd = 100,
nChr = 5,
Ne = ancient_decrease$Ne[1],
histGen = ancient_decrease$generations[-1],
histNe = ancient_decrease$Ne[-1]),
pop_migration = runMacs(nInd = 100,
nChr = 5,
manualCommand = migration_command,
manualGenLen = 1))
Both GONE and SNeP work with text files in Plink ped/map format. Look in the repository if you want to see the not too exciting details of how we extract 10 000 markers and save them to Plink format together with their positions.
GONE source code and binaries are found in their GitHub repository, which we can clone or simply download from the web. As I’m running on Linux, we will use the binaries in the Linux subfolder. Before doing anything else, we will have to set the permissions for each of the binaries stored in the PROGRAMMES directory, with chmod u+x to make them executable.
GONE consists of a set of binaries and a bash script that runs them in order. As the as the script assumes it’s always running from the directory where you put GONE and always writes the output files into the same directory, the easiest way to handle it is to duplicate the entire GONE directory for every run. This also duplicates the INPUT_PARAMETERS_FILE file that one would modify to change settings. Then, invoking GONE with default settings would be as simple as opening a terminal, moving to the working directory and running the script, feeding it the base name of the Plink file:
./script_GONE.sh pop_constant
Thus, we write a prep script like this that copies the entire folder, puts the data into it, and then runs GONE:
#!/bin/bash
## Gone needs all the content of the operating system-specific subfolder to be copied
## into a working directory to run from. Therefore we create the "gone" directory
## and copy in the Linux version of the software from the tools directory.
mkdir gone
cd gone
cp -r ../tools/GONE/Linux/* .
## Loop over all the cases and invoke the GONE runscript. Again, because GONE
## needs the data to be in the same directory, we copy the data files into the
## working directory.
for CASE in pop_constant pop_recent pop_ancient pop_migration pop_increase; do
cp ../simulation/${CASE}.* .
./script_GONE.sh ${CASE}
done
GONE puts its output files (named with the prefixes Output_Ne_, Output_d2_ and OUTPUT_ followed by the base name of the input files) in the same directory. The most interesting is Output_Ne_ which contains the estimates and the all caps OUTPUT_ file that contains logging information about the run.
Estimates look like this:
Ne averages over 40 independent estimates. Generation Geometric_mean 1 1616.29 2 1616.29 3 1616.29 4 1616.29 5 1291.22 6 1221.75 7 1194.16 8 1157.95 ...
And the log looks like this:
TOTAL NUMBER OF SNPs 10000 HARDY-WEINBERG DEVIATION -0.009012 Hardy-Weinberg deviation (sample) -0.003987 Hardy-Weinberg deviation (population) CHROMOSOME 1 NIND(real sample)=100 NSNP=2000 NSNP_calculations=2000 NSNP_+2alleles=0 NSNP_zeroes=0 NSNP_monomorphic=0 NIND_corrected=100.000000 freq_MAF=0.005000 F_dev_HW (sample)=-0.009017 F_dev_HW (pop)=-0.003992 Genetic distances available in map file ...
I will now discuss a couple of issues I ran into. Note, this should not be construed as any kind of criticism of the programs or their authors. Everyone is responsible for their own inability to properly read manuals; I just leave them here in case they are helpful to someone else.
DIVIDE .ped AND .map FILES IN CHROMOSOMES ./script_GONE.sh: line 96: ./PROGRAMMES/MANAGE_CHROMOSOMES2: Permission denied RUNNING ANALYSIS OF CHROMOSOMES ... cp: cannot stat 'chromosome*': No such file or directory bash: ./PROGRAMMES/LD_SNP_REAL3: Permission denied ...
DIVIDE .ped AND .map FILES IN CHROMOSOMES RUNNING ANALYSIS OF CHROMOSOMES ... CHROMOSOME ANALYSES took 0 seconds Running GONE Format error in file outfileLD Format error in file outfileLD Format error in file outfileLD Format error in file outfileLD Format error in file outfileLD ...
SNeP is only available as binaries on its Sourceforge page. Again, I’m using Linux binaries, so I downloaded the Linux binary from there and put it into a tools folder. The binary can be run from any directory, controlling the settings with command line flags. This would run SNeP on one of our simulated datasets, using the Haldane mapping function and correction of linkage disequilibrium values for sample size; these seem to be reasonable defaults:
./SNeP1.1 -ped simulation/pop_constant.ped -out snep/pop_constant -samplesize 2 -haldane
Thus, we write a run script like this:
#!/bin/bash
## As opposed to GONE, SNeP comes as one binary that can run from any directory. We still create
## a working directory to keep the output files in.
mkdir snep
## We loop over all cases, reading the data from the "simulation" directory,
## and directing the output to the "snep" directory. The settings are to
## correct r-squared for sample size using the factor 2, and to use the Haldane
## mapping function. We direct the output to a text file for logging purposes.
for CASE in pop_constant pop_recent pop_ancient pop_migration pop_increase; do
./tools/snep/SNeP1.1 \
-ped simulation/${CASE}.ped \
-out snep/${CASE} \
-samplesize 2 \
-haldane > snep/${CASE}_out.txt
done
SNeP creates two ouptut files with the given prefix, one with the extension .NeAll with the estimates a log file with the suffix SNeP.log file. Above, we also save the standard output to another log file.
Estimates look like this:
GenAgo Ne dist r2 r2SD items 13 593 3750544 0.0165248 0.0241242 37756 15 628 3272690 0.017411 0.0256172 34416 18 661 2843495 0.0184924 0.0281098 30785 20 681 2460406 0.0200596 0.0310288 27618 24 721 2117017 0.0214468 0.0313662 24898 ...
Issues I ran into:
/tools/snep/SNeP1.1 -ped constant.ped -out snep_constant ************************************* * SNeP * * v1.1 * * barbatom@cardiff.ac.uk * ************************************* Sat Dec 25 13:09:38 2021 The -out path provided gives an error, aborting.
Let’s read the results and look at the estimates!

Estimates from GONE, with default settings, and SNeP, with reasonable setting used in the SNeP paper, applied to simulated data from different scenarios. Grey dots are estimates, and black lines the true simulated population history. The estimates go on for a while, but as per the GONE paper’s recommendations, we should not pay attention to estimates further back in time where these methods are not accurate. That is, we should probably concentrate on the first 100 generations or so.
Again, this isn’t a systematic methods evaluation, so this shouldn’t be taken as a criticism of the programs or methods. But we can note that in these circumstances, the estimates capture some features of the simulated population histories but gets other features quite wrong. GONE estimates a recent decrease in the scenario with a recent decrease, but not the further decreases before, and a recent increase when there was a recent increase, but overestimates its size by a few thousand. In the migration scenario, GONE shows the kind of artefact the authors tell us to expect, namely a drastic population size drop. Unexpectedly, though, it estimates a similar drastic drop in the scenario with constant population size. SNeP captures the recent decrease, but underestimates its size. In fact, SNeP estimates a similar shape in all scenarios, both for increased, decreased and constant populations.
The plotting code looks something like this (see GitHub for the details): we create the file names, read all the output files into the same data frame with map_dfr, label them by what case they belong to by joining with the data frame of labels (with inner_join from dplyr) and make a plot with ggplot2. The true_descriptions data frame contains the true population histories used to simulate the data.
library(ggplot2)
library(readr)
cases <- tibble(case = c("pop_constant",
"pop_recent",
"pop_ancient",
"pop_migration",
"pop_increase"),
description = c("Constant",
"Recent decrease",
"Ancient decrease",
"Recent decrease with migration",
"Recent increase"))
snep_file_names <- paste("snep/", cases$case, ".NeAll", sep = "")
names(snep_file_names) <- cases$case
snep <- map_dfr(snep_file_names, read_tsv, .id = "case")
snep_descriptions <- inner_join(snep, cases)
snep_descriptions$description <- factor(snep_descriptions$description,
levels = cases$description)
## Make both a plot of the entire range of estimates, and a plot of the
## first 200 generations, which is the region where estimates are expected
## to be of higher quality
plot_snep_unconstrained <- ggplot() +
geom_point(aes(x = GenAgo, y = Ne),
data = snep_descriptions,
colour = "grey") +
facet_wrap(~ description,
scale = "free_y",
ncol = 2) +
geom_segment(aes(x = start,
y = Ne,
xend = end,
yend = Ne),
data = true_descriptions) +
theme_bw() +
theme(panel.grid = element_blank(),
strip.background = element_blank()) +
xlab("Generations ago")
plot_snep <- plot_snep_unconstrained +
coord_cartesian(xlim = c(0, 200), ylim = c(0, 3000))
When the results of a method don’t agree with the parameters of simulated data, the problem can either lie with the method or with the simulated data. And in this case, coalescent simulation is known to have problems with linkage disequilibrium. Here is a figure (Fig A, of appendix 2) of Nelson et al. (2020) who study the problems with coalescent simulations over long regions (such as the ones we simulated here).

The problem occurs for variants that are far apart (e.g. at around 100 = 1 expected recombinations between loci), where there should still be linkage disequilibrium, whereas the coalescent simulator (in this figure, ”msprime (Hudson)”) gives too low linkage disequilibrium. When we try to estimate effective population size late in population history, the methods rely on linkage equilibrium between markers far apart, and if they have too low linkage disequilibrium, the estimated population size should be too large. This does not appear to be what is going on here, but there might be more subtle problems with the simulated linkage disequilibrium that fools these methods; we could try something like Nelson et al.’s hybrid method or a forward simulation instead.
Literature
Barbato, M., Orozco-terWengel, P., Tapio, M., & Bruford, M. W. (2015). SNeP: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Frontiers in genetics, 6, 109.
Nelson, D., Kelleher, J., Ragsdale, A. P., Moreau, C., McVean, G., & Gravel, S. (2020). Accounting for long-range correlations in genome-wide simulations of large cohorts. PLoS genetics, 16(5), e1008619.
Santiago, E., Novo, I., Pardiñas, A. F., Saura, M., Wang, J., & Caballero, A. (2020). Recent demographic history inferred by high-resolution analysis of linkage disequilibrium. Molecular Biology and Evolution, 37(12), 3642-3653.
Let’s talk about something fun: the Advent of Code, where each day from the 1st to the 25th of December a new programming puzzle is presented for you to solve. A friend roped me into trying it this year, and I decided to try it in Python for learning purposes. Now halfway in, the difficulty level of the puzzles has increased, and I’m probably not doing all of them. Time to write down some reflections!
First, as one might expect, I have learned more Python this December than in several years of thinking ”I ought to learn a little Python at some point”. I’ve also started liking Python much more. Most of my professional Python experiences have been about running other peoples’ research code — sometimes with relative ease, and sometimes with copious dependence management. Dependence problems don’t make any language seem appealing. Advent of Code, on the other hand, is exactly what makes programming fun: small self-contained problems with well-defined solutions, test cases already prepared, and absolutely no need to install a years-old version of scikit-learn. And there is a lot to like about Python: list comprehensions (they are just fabulous!), automatic unpacking of tuples (yum!), numpy and pandas.
Second, it’s fun to notice what my most common errors are. I am a committed and enthusiastic R user, and you can tell from my errors: for the first few puzzles, I tended to accidentally write functions that returned None, because I unthinkingly relied on the R convention that the last expression in a function is automatically returned. The second most common error, frustratingly, is IndentationError: unexpected indent from having empty lines in my functions that are not indented. See, I like having whitespace between ”paragraphs” of code. While I’ve gotten over my gripes with some other common Python features, it still puzzles me that anyone would think that’s it’s a good idea to demand empty lines to be correctly indented. Even more puzzling, popular programmer’s text editor Atom, in its default configuration, deletes ”unncessary” whitespaces upon saving a Python source file.
After those, as expected, I’ve been making off-by-one errors like I was querying the UCSC Genome Browser for the first time (see this beautiful Biostars post for an explanation of that somewhat niche joke). I knew that Python counts from 0 where R counts from 1, but the difficulties don’t stop there. You also need to think about slices and ranges.
This gives you the first two elements of a list in Python:
pylist = ["a", "b", "c", "d"] pylist[0:2]
This gives you the first two elements of a list in R:
rlist <- list("a", "b", "c", "d")
rlist[1:2]
That is, 1:2 in R includes the last element, 2, whereas 0:2 in Python doesn’t. This is well known, well documented, mentioned in every tutorial — I still get it wrong.
When we add ranges that go backwards, like in this function from Day 5, and you can see where this poor R user needs to scratch his head (and write more tests):
def get_range(start, end): if (start > end): return range(start, end - 1, -1) else: return range(start, end + 1)
When I wrote about my common errors on Twitter, fellow quantitative geneticist Lorena Batista warned me about Python’s assignment, which works very differently from R’s. She was right. This has bitten me already. The way assignment works in Python, always passing around references to objects unless explicitly told to copy them, does not fit my intuitions about assignment at all. I feel uneasy about it and how it interacts with mutability — here we try to write proper functions that don’t have strange side effects, and then we clobber the parameters of the function instead. This will take some getting used to if I’m going to use Python more seriously.
I don’t want to start any language quarrels, but even I see how Python feels cleaner than R in certain ways. Maybe getting ranges to go backwards doesn’t feel as natural, but at least you can expect the standard library to consistent case when naming things, whereas in R you will see model.matrix, pivot_longer, and stringsAsFactors in the same script (but the latter not for long, bye default factor conversion, you will not be missed). On the other hand, Python suffers from confusion about where the relevant functions can be found: some live as static methods in an object named like the module, some live in the objects themselves, and some are free-floating. In R, almost everything is free-floating, and if some package has objects with methods (like the SimParam class in AlphaSimR) you will remember because it’s the exception.
A nice insight from the Advent of Code is that parsing is more important than I thought. I don’t mean that parsing custom text file formats is annoying and time consuming, even if it is. What I mean is that the second part of parsing, after you’ve solved the immediate problem of getting data out of a file, is data modelling.
Take for example Day 12, a graph-related problem. If you have have the computational wherewithal to parse the map into an adjacency list, you are already well on your way to solving the problem.
Or, take my favourite problem so far, Day 8: Seven Segment Search. This involved unscrambling some numbers from an imagined faulty display.
The data looks like this example, and it’s not super important what it means, except that the words are scrambled digits, and that each row represents one set of observations:
be cfbegad cbdgef fgaecd cgeb fdcge agebfd fecdb fabcd edb | fdgacbe cefdb cefbgd gcbe edbfga begcd cbg gc gcadebf fbgde acbgfd abcde gfcbed gfec | fcgedb cgb dgebacf gc fgaebd cg bdaec gdafb agbcfd gdcbef bgcad gfac gcb cdgabef | cg cg fdcagb cbg
It’s always ten words, followed by the delimiter, followed by another four words. This looks a little like a data frame, and I’m accustomed to thinking about tabular data structures. Therefore, unthinking, I used pandas to read this into a data frame, which I then sliced into two.
import pandas
import numpy as np
data = pandas.read_table("day8.txt", sep = " ", header = None)
digits = data.iloc[:, :10]
output = data.iloc[:, 11:15]
That was good enough to easily solve the first part, which was about counting particular digits (i.e., words with particular numbers of characters in them):
def count_simple_digits(digits):
""" Count 1, 4, 7, 8 in a column of digits """
simple = [digit for digit in digits if len(digit) in [2, 4, 3, 7]]
return len(simple)
sum([count_simple_digits(output[column]) for column in output])
Then comes part two, which was trickier, but more rewarding. I was pretty proud about my matrix-based solution, but that’s not the point here; I’m skipping over the functions that contain the solution. The point is that when I came to applying them to the real data, I had to do it in a clunky and I dare say unpythonic way:
## Apply to actual data
sorted_digits = digits.apply(sort_digits, axis = 0)
sorted_outputs = output.apply(sort_digits, axis = 0)
decoded = []
for ix in range(digits.shape[0]):
segment_sum = np.sum(get_segments_shared(sorted_digits.iloc[ix]), axis = 1).tolist()
matched_digits = match_digits(segment_sum, segment_sums_normal)
decoded.append(decode_output(sorted_outputs.iloc[ix].tolist(), sorted_digits.iloc[ix].tolist(), matched_digits))
Look at that indexing over rows, which is not a smooth operation on a data frame. My problem here is that I stored the data on the same set of digits over ten columns (and four more columns for the words after the delimiter), when it would have been much more natural to have each data point pertaining to the same set of digits stored together in one structure — anything that you can easily iterate over without an indexed for loop.
The lesson, which applies to R code as well, is to not always reach for the tabular data structure just because it’s familiar and comes with a nice read_csv function, but to give more thought to data modelling.
It can be a touchy topic whether people who do science or make use of outcomes of science are supposed to believe in science. I can think of at least three ways that we may or may not believe in science.
Imagine that we try to apply the current state of scientific knowledge to some everyday occurrence. It could be something that happens when cooking, a very particular sensation you notice in your body, anything that you have practical experience-based knowledge about, one of those particular phenomena that anyone notices when in some practical line of work. Every trade has its own specialised terminology and models of explanation that develops based on experience.
When looking closely, we will often find that the current state of scientific knowledge does not have a theory, model or explanation ready for us. This might not be because the problem is so hard to be unsolvable. It might be because the problem has simply been overlooked.
That means that if we want to maintain a world view where science has the answer, we don’t just need to believe in particular statements that science makes. We also need to trust that a scientific explanation can be developed for many cases where one doesn’t exist yet.
I can’t remember who I read who used this image (I think it might have been a mathematician): knowledge is not a solid body of work, but more like tunnels dug out of the mostly unknown. We don’t know most things, but we know some paths dug through this mass.
There is another popular saying: the good thing about it is that it works whether you believe in it or not — said by Neil DeGrasse Tyson about science and attributed to Niels Bohr about superstition.
This seems to be true, not just about deep cases such as keeping one set of methodological assumptions for your scientific work and another, say a deeply spiritual religious one, for your personal life. It seems to be true about the way we expect scientists to work and communicate on an everyday level. Scientists do not need to be committed to every statement in their writing or every hypothesis they pursue.
Dang & Bright (2021) ask what demands we should put on individual scientific contributions (e.g. journal articles). They consider assertion norms, the idea that certain types of utterances are appropriate for a certain context, and argue that scientific contributions should not be held to these norms. That is, scientific claims need not be something the researcher knows, has justification for, or believes.
They give an example of a scientist who comes up with a new hypothesis, and performs some research that supports it. Overall, however, the literature does not support it. The scientist publishes a paper advocating for the hypothesis. In a few years, further research demonstrates that it is false. They suggest that in this case, the scientist has done what we expect scientists to do, even if:
They try to explain these norms of communication with reference to how the scientific community learns. They argue that this way of communicating facilitates an intellectual division of labour: different researchers consider different ideas and theories, that might at times be at odds, and therefore there is value to allowing them to them saying conflicting things.
The rejected norms were picked as the basis of our inquiry since they had been found plausible or defensible as norms for assertion, which is at least a somewhat related activity of putting forward scientific public avowals. So why this discrepancy? In short, we think this is because the social enterprise of inquiry requires that we allow people to be more lax in certain contexts than we normally require of individuals offering testimony, and through a long process of cultural evolution the scientific community developed norms of avowal to accommodate that fact.
Another reason, pragmatically, why scientists might not be fully committed to everything they’ve written is that, at least in natural science, writing often happens by committee. And not just a committee of co-authors, but also by reviewers and editors, who also take part in negotiations about what can be claimed in any piece of peer reviewed writing. Yes, there are people who like to say that every co-author needs to be able to take full responsibility for everything written — but the real lowest common denominator seems that none of the co-authors objects too much to what is written.
Finally, there appears to be two ways to think about claims when reading or writing scientific papers, that may explain that researchers can be at the same time highly critical of some claims, and somewhat credulous about other claims. Think of your typical introduction to an empirical paper, which will often have citations in passing to stylised facts that are taken to be true, but not explored at any depth — those ”neutral citations” that some think are shallow and damaging. Does that mean that scientists are lazy about the truth, just believing the first thing they see and jumping on citation bandwagons? Maybe. Because there is no other way they could write, really.
When we read or write about science, we have to divide claims into those that are currently under critical scrutiny, and those that are currently considered background. If we believe Quine, there isn’t an easy way to separate these two types of ideas, no logical division into hypothesis proper and auxiliary assumption. But we still need to make it, possibly in an arbitrary way, unless we want our research project to be an endless rabbit hole of questions. This does not mean that we have to believe the background claims in any deeper sense. They might be up for scrutiny tomorrow.
This is another reason why it’s a bad idea to try to learn a field by reading the introductions to empirical papers. Introductions are not critical expositions that synthesise evidence and give the best possible view of the state of the field. They just try to get the important background out of the way so we can move on with the work at hand.
Literature
Dang, H., & Bright, L. K. (2021). Scientific conclusions need not be accurate, justified, or believed by their authors. Synthese.
Philip Ball, who is a knowledgeable and thoughtful science writer, published an piece in the Guardian a couple of months ago about the misunderstood legacy of the human genome project: ”20 years after the human genome was first sequenced, dangerous gene myths abound”.
The human genome project published the draft reference genome for the human species 20 years ago. Ball argues, in short, that the project was oversold with promises that it couldn’t deliver, and consequently has not delivered. Instead, the genome project was good for other things that had more to do with technology development and scientific infrastructure. The sequencing of the human genome was the platform for modern genome science, but it didn’t, for example, cure cancer or uncover a complete set of instructions for building a human.
He also argues that the rhetoric of human genome hype, which did not end with the promotion of the human genome project (see the ENCODE robot punching cancer in the face, for example), is harmful. It is scientifically harmful because it oversimplifies modern genetics, and it is politically harmful because it aligns well with genetic determinism and scientific racism.
I believe Ball is entirely right about this.
The breathless hype around the human genome project was embarrassing. Ball quotes some fragments, but you can to to the current human genome project site and enjoy quotes like ”it’s a transformative textbook of medicine, with insights that will give health care providers immense new powers to treat, prevent and cure disease”. This image has some metonymical truth to it — human genomics is helping medical science in different ways — but even as a metaphor, it is obviously false. You can go look at the human reference genome if you want, and you will discover that the ”text” such as it is looks more like this than a medical textbook:
TTTTTTTTCCTTTTTTTTCTTTTGAGATGGAGTCTCGCTCTGCCGCCCAGGCTGGAGTGC
AGTAGCTCGATCTCTGCTCACTGCAAGCTCCGCCTCCCGGGTTCACGCCATTCTCCTGCC
TCAGCCTCCTGAGTAGCTGGGACTACAGGCGCCCACCACCATGCCCAGCTAATTTTTTTT
TTTTTTTTTTTGGTATTTTTAGTAGAGACGGGGTTTCACCGTGTTAGCCAGGATGGTCTC
AATCTCCTGACCTTGTGATCCGCCCGCCTCGGCCTCCCACAGTGCTGGGATTACAGGC
This is a human Alu element from chromosome 17. It’s also in an intron of a gene, flanking a promoter, a few hundred basepairs away from an insulator (see the Ensembl genome browser) … All that is stuff that cannot be read from the sequence alone. You might be able to tell that it’s Alu if you’re an Alu genius or run a sequence recognition software, but there is no to read the other contextual genomic information, and there is no way you can tell anything about human health by reading it.
I think Ball is right that this is part of simplistic genetics that doesn’t appreciate the complexity either quantitative or molecular genetics. In short, quantitative genetics, as a framework, says that inheritance of traits between relatives is due to thousands and thousands of genetic differences each of them with tiny effects. Molecular genetics says that each of those genetic differences may operate through any of a dizzying selection of Rube Goldberg-esque molecular mechanisms, to the point where understanding one of them might be a lifetime of laboratory investigation.
Simple inheritance is essentially a fiction, or put more politely: a simple model that is useful as a step to build up a more better picture of inheritance. This is not knew; the knowledge that everything of note is complex has been around since the beginning of genetics. Even rare genetic diseases understood as monogenic are caused by sometimes thousands of different variants that happen in a particular small subset of the genome. Really simple traits, in the sense of one variant–one phenotype, seldom happen in large mixing and migrating populations like humans; they may occur in crosses constructed in the lab, or in extreme structured populations like dog breeds or possibly with balancing selection.
Ball is also right about what it was most useful about the human genome project: it enabled research at scale into human genetic variation, and it stimulated development of sequencing methods, both generating and using DNA sequence. Lowe (2018) talks about ”thick” sequencing, a notion of sequencing that includes associated activities like assembly, annotation and distribution to a community of researchers — on top of ”thin” sequencing as determination of sequences of base pairs. Thick sequencing better captures how genome sequencing is used and stimulates other research, and aligns with how sequencing is an iterative process, where reference genomes are successively refined and updated in the face of new data, expert knowledge and quality checking.
It is hard to imagine gene editing like CRISPR being applied in any human cell without a good genome sequence to help find out what to cut out and what to put instead. It is hard to imagine the developments in functional genomics that all use short read sequencing as a read-out without having a good genome sequence to anchor the reads on. It is possible to imagine genome-wide association just based on very dense linkage maps, but it is a bit far-fetched. And so on.
Now, this raises a painful but interesting question: Would the genome project ever have gotten funded on reasonable promises and reasonable uncertainties? If not, how do we feel about the genome hype — necessary evil, unforgivable deception, something in-between? Ball seems to think that gene hype was an honest mistake and that scientists were surprised that genomes turned out to be more complicated than anticipated. Unlike him, I do not believe that most researchers honestly believed the hype — they must have known that they were overselling like crazy. They were no fools.
An example of this is the story about how many genes humans have. Ball writes:
All the same, scientists thought genes and traits could be readily matched, like those children’s puzzles in which you trace convoluted links between two sets of items. That misconception explains why most geneticists overestimated the total number of human genes by a factor of several-fold – an error typically presented now with a grinning “Oops!” rather than as a sign of a fundamental error about what genes are and how they work.
This is a complicated history. Gene number estimates are varied, but enjoy this passage from Lewontin in 1977:
The number of genes is not large
While higher organisms have enough DNA to specify from 100,000 to 1,000,000 proteins of average size, it appears that the actual number of cistrons does not exceed a few thousand. Thus, saturation lethal mapping of the fourth chromosome (Hochman, 1971) and the X chromosome (Judd, Shen and Kaufman, 1972) of Drosophila melanogbaster make it appear that there is one cistron per salivary chromosome band, of which there are 5,000 in this species. Whether 5,000 is a large or small number of total genes depends, of course, on the degree of interaction of various cistrons in influencing various traits. Nevertheless, it is apparent that either a given trait is strongly influenced by only a small number of genes, or else there is a high order of gene interactions among developmental systems. With 5,000 genes we cannot maintain a view that different parts of the organism are both independent genetically and each influenced by large number of gene loci.
I don’t know if underestimating by an few folds is worse than overestimating by a few folds (D. melanogaster has 15,000 protein-coding genes or so), but the point is that knowledgeable geneticists did not go around believing that there was a simple 1-to-1 mapping between genes and traits, or even between genes and proteins at this time. I know Lewontin is a population geneticist, and in the popular mythology population geneticists are nothing but single-minded bean counters who do not appreciate the complexity of molecular biology … but you know, they were no fools.
One thing Ball gets wrong is evolutionary genetics, where he mixes genetic concepts that, really, have very little to do with each other despite superficially sounding similar.
Yet plenty remain happy to propagate the misleading idea that we are “gene machines” and our DNA is our “blueprint”. It is no wonder that public understanding of genetics is so blighted by notions of genetic determinism – not to mention the now ludicrous (but lucrative) idea that DNA genealogy tells you which percentage of you is “Scots”, “sub-Saharan African” or “Neanderthal”.
This passage smushes two very different parts of genetics together, that don’t belong together and have nothing to do with with the preceding questions about how many genes there are or if the DNA is a blueprint: The gene-centric view of adaptation, a way of thinking of natural selection where you imagine genetic variants (not organisms, not genomes, not populations or species) as competing for reproduction; and genetic genealogy and ancestry models, where you describe how individuals are related based on the genetic variation they carry. The gene-centric view is about adaptation, while genetic genealogy works because of effectively neutral genetics that just floats around, giving us a unique individual barcode due to the sheer combinatorics.
He doesn’t elaborate on the gene machines, but it links to a paper (Ridley 1984) on Williams’ and Dawkins’ ”selfish gene” or ”gene-centric perspective”. I’ve been on about this before, but when evolutionary geneticists say ”selfish gene”, they don’t mean ”the selfish protein-coding DNA element”; they mean something closer to ”the selfish allele”. They are not committed to any view that the genome is a blueprint, or that only protein-coding genes matter to adaptation, or that there is a 1-to-1 correspondence between genetic variants and traits.
This is the problem with correcting misconceptions in genetics: it’s easy to chide others for being confused about the parts you know well, and then make a hash of some other parts that you don’t know very well yourself. Maybe when researchers say ”gene” in a context that doesn’t sound right to you, they have a different use of the word in mind … or they’re conceptually confused fools, who knows.
Literature
Lewontin, R. C. (1977). The relevance of molecular biology to plant and animal breeding. In International Conference on Quantitative Genetics. Ames, Iowa (USA). 16-21 Aug 1976.
Lowe, J. W. (2018). Sequencing through thick and thin: Historiographical and philosophical implications. Studies in History and Philosophy of Science Part C: Studies in History and Philosophy of Biological and Biomedical Sciences, 72, 10-27.



