Genuttryck och höns är ju två av mina intressen, så jag blev givetvis intresserad av Genetic architecture of gene expression in the chicken av Stanley m fl i BMC Genomics. Genetisk arkitektur brukar betyda information om genetiska varianter bakom någon egenskap, men de använder tydligen uttrycket på ett annat sätt. Vad de gjort är ett nätverk som visar korrelationerna i genuttryck mellan 8650 hönsgener, byggt på ungefär 1000 publicerade mikroarrayresultat. Det blir en rätt snygg illustration och en massa öppna frågor. Först något om viktade genuttrycksnätverk!
Metoden de använder kallas WGCNA (det finns ett gäng artiklar om den, se till exempel Langfelder & Horvath 2008 om implementationen i R). Idén är att beskriva hur gener hänger samman genom att se hur deras uttrycksnivåer korrelerar. Det första steget är att göra en stor korrelationsmatris. Så, vilka korrelationer är stora nog att vara intressanta? Istället för att dra en gräns (säg korrelationer större än 0.5 är intressanta) så viktas de med en potensfuktion. Varför en potensfunktion? Jo många nätverk, både biologiska och andra, har det som kallas skalfri struktur där bågarna är fördelade enligt just en potensfunktion. Om nätverket viktas med rätt potens blir det typ skalfritt. En gör alltså antagandet att små korrelationer i allmänhet är oviktiga, men framhäver de stora ännu mer.
Det är god ton bland folk som använder mikroarrayer (”genchip”) och ett krav från många tidskrifter att mikroarraydata publiceras i någon av de offentliga databaserna för sådana. Därför finns det mängder av råa genuttryckdata på internet för den som vill laborera med dem. Det har författarna dragit nytta av och laddat hem varenda hönsarray (av ett visst märke, Affymetrix) de kunnat hitta. Det blev totalt dryga 1000 chip från 67 publicerade experiment på olika vävnader från olika höns. De tog inga hänsyn till under vilka förhållanden genuttrycksvärdena samlades in i första rummet, utan justerade bara för systematiska skillnader mellan experiment.
WGCNA har också ett sätt att dela upp nätverket i moduler (för nördarna där ute: det är hierarkisk klustring, en algoritm för att dela upp det resulterande trädet i grupper och sammanslagning av moduler som är nära varandra). Efter att ha delat upp sitt nätverk i moduler testade de modulerna för anrikning av gener med olika funktioner (”har modulen fler gener av den här typen än det borde bli om de vore slumpvis fördelade”). Här är en Cytoscape-bild av nätverket: varje nod är en gen och varje färg en modul. Mellan noderna går bågar som var och en har en tillhörande vikt. Pilarna pekar ut anrikade funktioner.
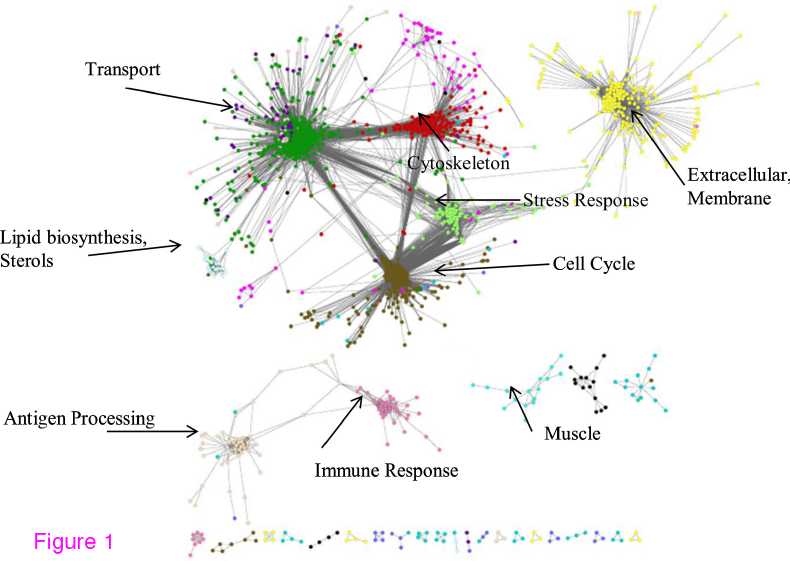
(Figur 1, Stanley et al 2013)
Det blir ju en snygg bild och kanske inte så oväntat att antigen processing och immune response som båda har med immunförsvaret att göra eller cytoskelettet och cellcykeln hamnar nära varandra. För min del undrar jag över några saker som inte definieras i artikeln — exakt vad menar de med att en nod är ”among the most highly connected” eller att en modul har ”little or no connections to the rest of the network”. WGCNA har ingen tröskel för när två noder inte anses kopplade, bara bågar med väldigt små vikter. När anser de att en vikt är tillräckligt liten för att inte finnas? En kan lägga märke till att det som pekas ut i diagrammet ovan är ganska allmängiltiga biologiska funktioner. En kan fråga sig om det skulle finnas några skillnader hönsnätverk, ett människonätverk och ett jästnätverk. Går det alls att se några hönsspecifika detaljer? Det finns genuttrycksnätverk som korsar artgränserna, undrar hur mycket det skiljer sig från det här.
Litteratur
Dragana Stanley, Nathan S Watson-Haigh, Christopher JE Cowled, Robert J Moore. (2013) Genetic architecture of gene expression in the chicken. BMC Genomics 14 doi:10.1186/1471-2164-14-13
Peter Langfelder, Steve Horvath. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9 doi:10.1186/1471-2105-9-559







{kind=link}
{kind=link}
{kind=link}
{kind=link}