This preprint was posted on bioRxiv and Haldane’s sieve. It tells the story of one of the best known genetic variants affecting behaviour, the foraging gene in Drosophila melanogaster. for is still a nice example of a large-effect variant causing (developmentally) pleiotropic effects. However, Turner & al present evidence questioning whether for has any substantial effect in natural populations of flies. I think it’s self-evident why I’m interested.
They look at previous evidence for foraging as a quantitative trait gene in files sampled from natural populations and perform genome-wide association and population genetic tests with 35 DGRP lines, finding nothing at the for locus.
Comments:
(Since this is a preprint, I will feel free to suggest what I think could be improvements to the manuscript. Obviously, these are just my opinions.)
I’m not convinced one can really separate a unimodal from a bimodal distribution with 36 data points? Below are a few histograms simulated from a mixture of two normal distributions where 25 samples are ”rovers” and 11 ”sitters”.
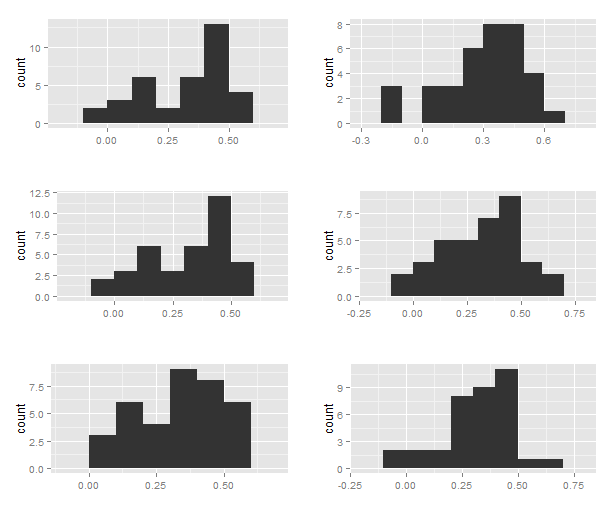
For fun, I also tested for normality with the Shapiro-Wilks’ test as the authors did, and about half of 1000 tests reject. My histograms should not be overinterpreted; I just generated two normal distributions with means log10(2.66) and log10(1.3) with standard deviations 0.1. I don’t know the actual standard deviations of the forS and forR reference strains. Of course, when the standard deviation is small enough, the distributions clearly separate and Shapiro-Wilks’ test will reject.
Power is difficult, but in this case the authors are looking at a well-known effect. They should be able to postulate some reasonable effect-sizes given the literature and the difference between the reference strains and make sure that they’re actually powered to detect it. 35 individuals for a GWAS is not much. They may still have good power to detect a effect of the size expected at for, at least in the single-point test, but it would be nice to demonstrate it. Power feels particularly pertinent as the authors claim to find evidence of absence. The same thing should apply to the population genetic tests, though it’s probably harder to know what effects to expect there.
The authors discuss alternative interpretations, and mention the fact that in their hands the reference strains did not travel nearly as long as in previous experiments. How likely is it, though, that the variant isn’t segregating in Raleigh but in the populations previously sampled?
Literature
Thomas Turner, Christopher C Giauque, Daniel R Schrider, Andrew D Kern. (2014) Genome-wide association of foraging behavior in Drosophila melanogaster fails to support large-effect alleles at the foraging gene. Preprint on bioaRxiv. doi: 10.1101/004325

Thanks for your interest in our manuscript! I agree that our data are hardly a death-knell for the ”common large-effect variant hypothesis” at foraging. Power is certainly an issue. However, I would suggest that our data are stronger than published datasets that claim to support this hypothesis. Add to that the publication bias against negative results, and we thought it was very important to publish our results and compare them to the existing literature. In my view, the foundations of this ”classic” story seem surprisingly shaky. If our manuscript results in better data coming to light that support the common variant hypothesis, that would be terrific!
Regards,
Thomas Turner
And thank you for commenting!
I thought this was really interesting! I’m all for revisiting paradigmatic genes, experiments and systems, and forager is particularly interesting because of the supposed balancing selection.
And of course, by bringing up power, I’m not suggesting the experiment shouldn’t be done or published. This seems like one of those cases where it’s possible to look at power to help interpretation. And as Gelman writes, one never has a large enough N, because if one goes out and gets a larger sample size there’ll always be more predictors and interactions to fit.
Cheers,
martin
I have posted some comments about the analysis of this paper over on biorxiv, with the analysis, code and data on github.
http://default/content/early/2014/04/20/004325#comment-1362483140
I saw the link at bioaRxiv earlier, but I’ve just skimmed it so far. The comparison between labs is interesting, since it opens up to getting at gene by environment interactions.
Cheers,
m.