Genetics is a data science, right?
One of my Summer of data science learning points was to play with out of the box prediction tools. So let’s try out a few genomic prediction methods. The code is on GitHub, and the simulated data are on Figshare.
Genomic selection is the happy melding of quantitative and molecular genetics. It means using genetic markers en masse to predict traits and and make breeding decisions. It can give you better accuracy in choosing the right plants or animals to pair, and it can allow you to take shortcuts by DNA testing individuals instead of having to test them or their offspring for the trait. There are a bunch of statistical models that can be used for genomic prediction. Now, the choice of prediction algorithm is probably not the most important part of genomic selection, but bear with me.
First, we need some data. For this example, I used AlphaSim (Faux & al 2016), and the AlphaSim graphical user interface, to simulate a toy breeding population. We simulate 10 chromosomes á 100 cM, with 100 additively acting causal variants and 2000 genetic markers per chromosome. The initial genotypes come from neutral simulations. We run one generation of random mating, then three generations of selection on trait values. Each generation has 1000 individuals, with 25 males and 500 females breeding.
So we’re talking a small-ish population with a lot of relatedness and reproductive skew on the male side. We will use the two first two generations of selection (2000 individuals) to train, and try to predict the breeding values of the fourth generation (1000 individuals). Let’s use two of the typical mixed models used for genomic selection, and two tree methods.
We start by splitting the dataset and centring the genotypes by subtracting the mean of each column. Centring will not change predictions, but it may help with fitting the models (Strandén & Christensen 2011).
Let’s begin with the workhorse of genomic prediction: the linear mixed model where all marker coefficients are drawn from a normal distribution. This works out to be the same as GBLUP, the GCTA model, GREML, … a beloved child has many names. We can fit it with the R package BGLR. If we predict values for the held-out testing generation and compare with the real (simulated) values, it looks like this. The first panel shows a comparison with phenotypes, and the second with breeding values.
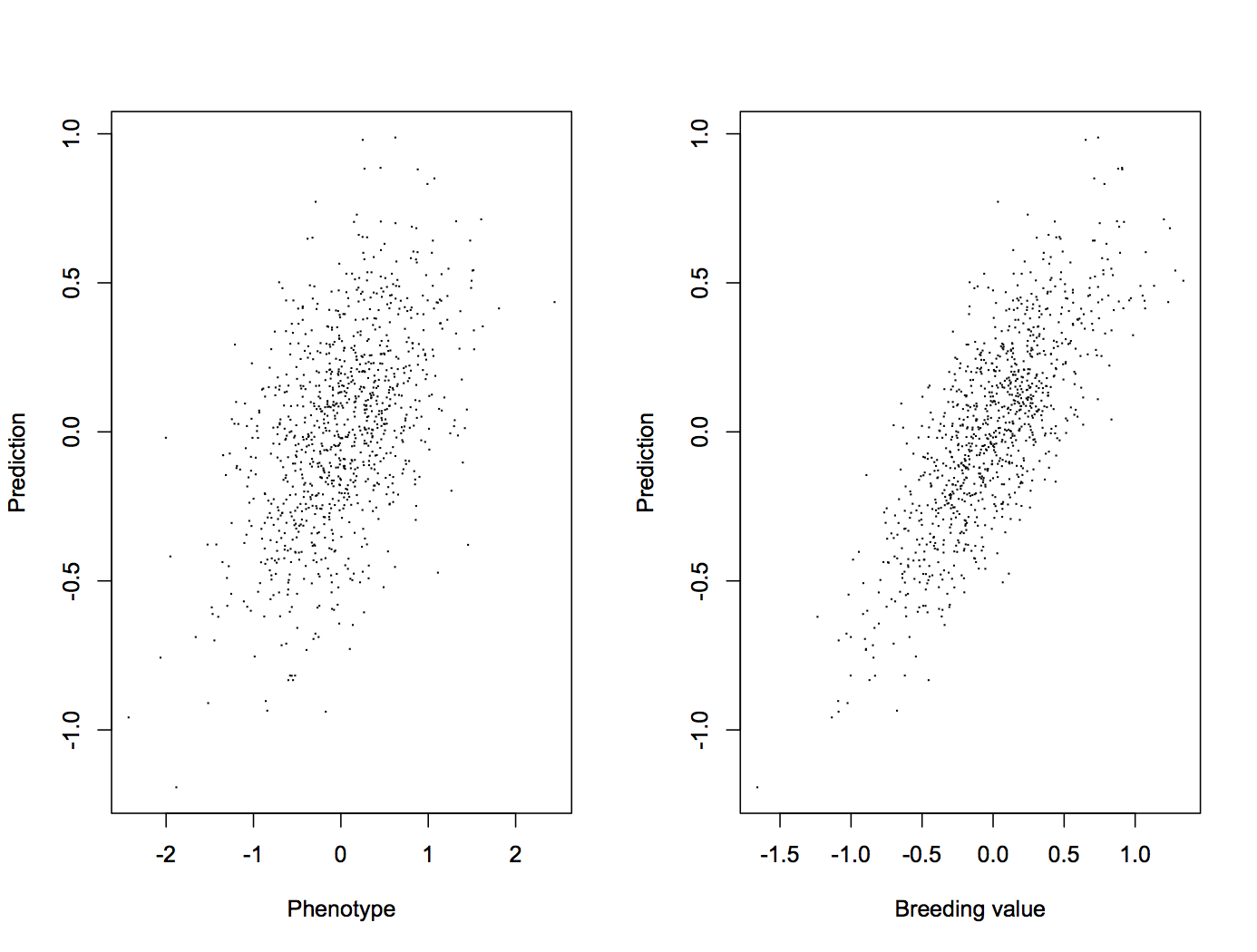
This gives us correlations of 0.49 between prediction and phenotype, and 0.77 between prediction and breeding value.
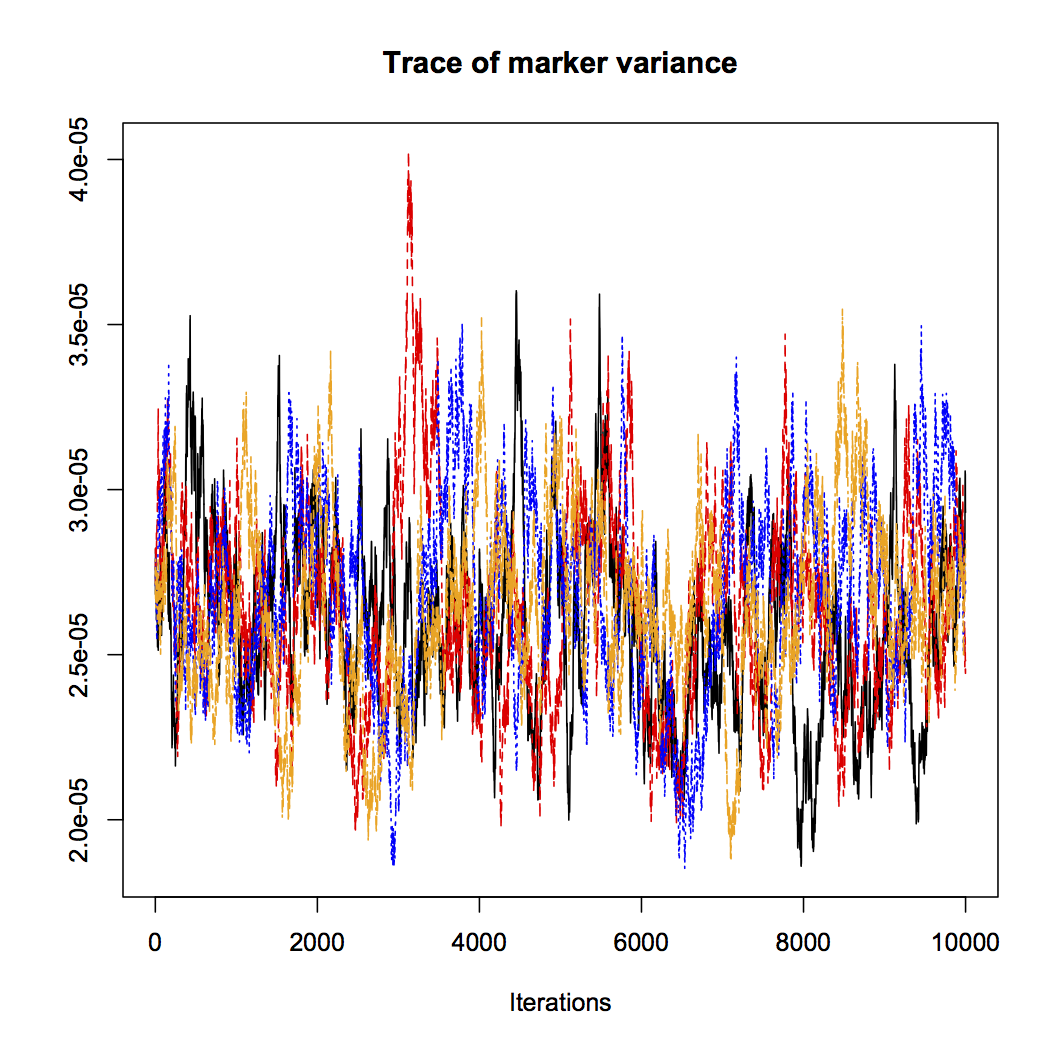
This is a plot of the Markov chain Monte Carlo we use to sample from the model. If a chain behaves well, it is supposed to have converged on the target distribution, and there is supposed to be low autocorrelation. Here is a trace plot of four chains for the marker variance (with the coda package). We try to be responsible Bayesian citizens and run the analysis multiple times, and with four chains we get very similar results from each of them, and a potential scale reduction factor of 1.01 (it should be close to 1 when it works). But the autocorrelation is high, so the chains do not explore the posterior distribution very efficiently.
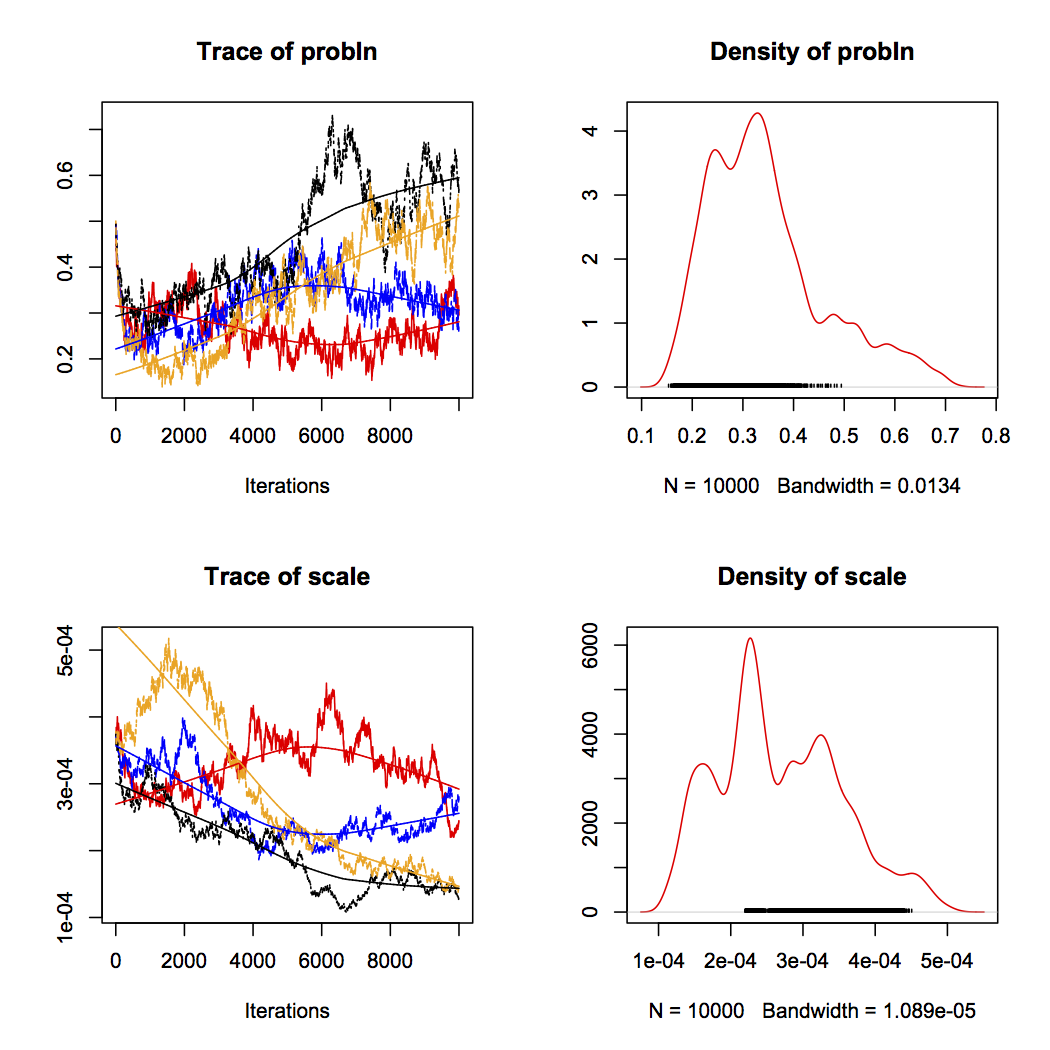
BGLR can also fit a few of the ”Bayesian alphabet” variants of the mixed model. They put different priors on the distribution of marker coefficients allow for large effect variants. BayesB uses a mixture prior, where a lot of effects are assumed to be zero (Meuwissen, Hayes & Goddard 2001). The way we simulated the dataset is actually close to the BayesB model: a lot of variants have no effect. However, mixture models like BayesB are notoriously difficult to fit — and in this case, it clearly doesn’t work that well. The plots below show chains for two BayesB parameters, with potential scale reduction factors of 1.4 and 1.5. So, even if the model gives us the same accuracy as ridge regression (0.77), we can’t know if this reflects what BayesB could do.
On to the trees! Let’s try Random forest and Bayesian additive regression trees (BART). Regression trees make models as bifurcating trees. Something like the regression variant of: ”If the animal has a beak, check if it has a venomous spur. If it does, say that it’s a platypus. If it doesn’t, check whether it quacks like a duck …” The random forest makes a lot of trees on random subsets of the data, and combines the inferences from them. BART makes a sum of trees. Both a random forest (randomForest package) and a BART model on this dataset (fit with bartMachine package), gives a lower accuracy — 0.66 for random forest and 0.72 for BART. This is not so unexpected, because the strength of tree models seems to lie in capturing non-additive effects. And this dataset, by construction, has purely additive inheritance. Both BART and random forest have hyperparameters that one needs to set. I used package defaults for random forest, values that worked well for Waldmann (2016), but one probably should choose them by cross validation.
Finally, we can use classical quantitative genetics to estimate breeding values from the pedigree and relatives’ trait values. Fitting the so called animal model in two ways (pedigree package and MCMCglmm) give accuracies of 0.59 and 0.60.
So, in summary, we recover the common wisdom that the linear mixed model does the job well. It was more accurate than just pedigree, and a bit better than BART. Of course, the point of this post is not to make a fair comparison of methods. Also, the real magic of genomic selection, presumably, happens on every step of the way. How do you get to that neat individual-by-marker matrix in the first place, how do you deal with missing data and data from different sources, what and when do you measure, what do you do with the predictions … But you knew that already.
