This paper (Wallace 2020) is about improvements to the colocalisation method for genome-wide association studies called coloc. If you have an association to trait 1 in a region, and another association with trait 2, coloc investigates whether they are caused by the same variant or not. I’ve never used coloc, but I’m interested because setting reasonable priors is related to getting reasonable parameters for genetic architecture.
The paper also looks at how coloc is used in the literature (with default settings, unsurprisingly), and extends coloc to relax the assumption of only one causal variant per region. In that way, it’s a solid example of thoughtfully updating a popular method.
(A note about style: This isn’t the clearest paper, for a few reasons. The structure of the introduction is indirect, talking a lot about Mendelian randomisation before concluding that coloc isn’t Mendelian randomisation. The paper also uses numbered hypotheses H1-H4 instead of spelling out what they mean … If you feel a little stupid reading it, it’s not just you.)
coloc is what we old QTL mappers call a pleiotropy versus linkage test. It tries to distinguish five scenarios: no association, trait 1 only, trait 2 only, both traits with linked variants, both traits with the same variant.
This paper deals with the priors: What is the prior probability of a causal association to trait 1 only 


They reparametrise the priors so that it becomes possible to get some estimates from the literature. They work with the probability that a SNP is causally associated with each trait (which means adding the probabilities of association 

How about
If traits were independent, you could just multiply 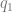
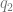
You could also look at the genetic correlation between traits. It makes sense that the overall genetic relationship between two traits should inform the prior that you see overlap at this particular locus. This gives a lower limit for
Attempts to colocalise disease and eQTL signals have ranged from underwhelming to positive. One key difference between outcomes is the disease-specific relevance of the cell types considered, which is consistent with variable chromatin state enrichment in different GWAS according to cell type. For example, studies considering the overlap of open chromatin and GWAS signals have convincingly shown that tissue relevance varies by up to 10 fold, with pancreatic islets of greatest relevance for traits like insulin sensitivity and immune cells for immune-mediated diseases. This suggests that
Literature
Wallace, Chris. ”Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses.” PLoS Genetics 16.4 (2020): e1008720.
