Cullen Roth posted a beautiful animation of quantitative trait locus mapping on Twitter. It is pretty amazing. I wanted to try to make something similar in R with gganimate. It’s not going to be as beautiful as Roth’s animation, but it will use the same main idea of showing both a test statistic along the genome, and the underlying genotypes and trait values. For example, Roth’s animation has an inset scatterplot that appears above the peak after it’s been reached; to do that I think we would have to go a bit lower-level than gganimate and place our plots ourselves.
First, we’ll look at a locus associated with body weight in chickens (with data from Henriksen et al. 2016), and then a simulated example. We will use ggplot2 with gganimate and a magick trick for combining the two animations. Here are some pertinent snippets of the code; as usual, find the whole thing on Github.
LOD curve
We will use R/qtl for the linkage mapping. We start by loading the data file (Supplementary Dataset from Henriksen et al. 2016). A couple of individuals have missing covariates, so we won’t be able to use them. This piece of code first reads the cross file, and then removes the two offending rows.
library(qtl)
## Read cross file
cross <- read.cross(format = "csv",
file = "41598_2016_BFsrep34031_MOESM83_ESM.csv")
cross <- subset(cross, ind = c("-34336", "-34233"))
For nice plotting, let’s restrict ourselves to fully informative markers (that is, the ones that tell the two founder lines of the cross apart). There are some partially informative ones in the dataset too, and R/qtl can get some information out of them thanks to genotype probability calculations with its Hidden Markov Model. They don’t make for nice scatterplots though. This piece of code extracts the genotypes and identifies informative markers as the ones that only have genotypes codes 1, 2 or 3 (homozygote, heterozygote and other homozygote), but not 5 and 6, which are used for partially informative markers.
## Get informative markers and combine with phenotypes for plotting
geno <- as.data.frame(pull.geno(cross,
chr = 1))
geno_values <- lapply(geno, unique)
informative <- unlist(lapply(geno_values,
function(g) all(g %in% c(1:3, NA))))
geno_informative <- geno[informative]
Now for the actual scan. We run a single QTL scan with covariates (sex, batch that the chickens were reared in, and principal components of genotypes), and pull out the logarithm of the odds (LOD) across chromosome 1. This piece of code first prepares a design matrix of the covariates, and then runs a scan of chromosome 1.
## Prepare covariates
pheno <- pull.pheno(cross)
covar <- model.matrix(~ sex_number + batch + PC1 + PC2 + PC3 + PC4 +
PC5 + PC6 + PC7 + PC8 + PC9 + PC10,
pheno,
na.action = na.exclude)[,-1]
scan <- scanone(cross = cross,
pheno.col = "weight_212_days",
method = "hk",
chr = 1,
addcovar = covar)
Here is the LOD curve along chromosome 1 that want to animate. The peak is the biggest-effect growth locus in this intercross, known as ”growth1”.
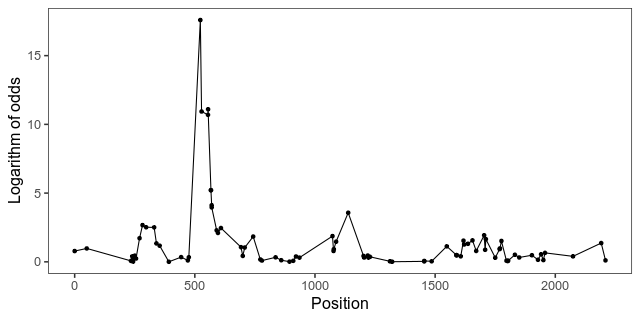
With gganimate, animating the points is as easy as adding a transition layer. This piece of code first makes a list of some formatting for our graphics, then extracts the LOD scores from the scan object, and makes the plot. By setting cumulative in transition_manual the animation will add one data point at the time, while keeping the old ones.
library(ggplot2)
library(gganimate)
formatting <- list(theme_bw(base_size = 16),
theme(panel.grid = element_blank(),
strip.background = element_blank(),
legend.position = "none"),
scale_colour_manual(values =
c("red", "purple", "blue")))
lod <- as.data.frame(scan)
lod <- lod[informative,]
lod$marker_number <- 1:nrow(lod)
plot_lod <- qplot(x = pos,
y = lod,
data = lod,
geom = c("point", "line")) +
ylab("Logarithm of odds") +
xlab("Position") +
formatting +
transition_manual(marker_number,
cumulative = TRUE)
Plot of the underlying data
We also want a scatterplot of the data. Here what a jittered scatterplot will look like at the peak. The horizontal axes are genotypes (one homozygote, heterozygote in the middle, the other homozygote) and the vertical axis is the body mass in grams. We’ve separated the sexes into small multiples. Whether to give both sexes the same vertical axis or not is a judgement call. The hens weigh a lot less than the roosters, which means that it’s harder to see patterns among them when put on the same axis as the roosters. On the other hand, if we give the sexes different axes, it will hide that difference.
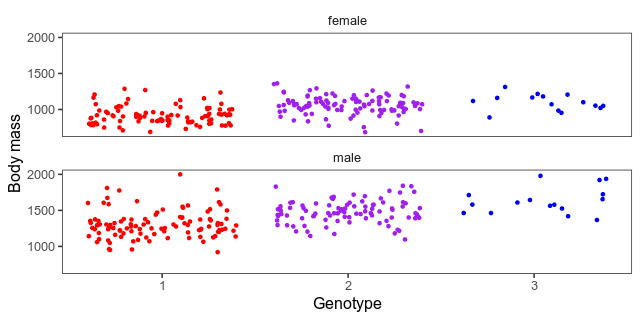
This piece of code builds a combined data frame with informative genotypes and body mass. Then, it makes the above plot for each marker into an animation.
library(tidyr)
## Combined genotypes and weight
geno_informative$id <- pheno$id
geno_informative$w212 <- pheno$weight_212_days
geno_informative$sex <- pheno$sex_number
melted <- pivot_longer(geno_informative,
-c("id", "w212", "sex"))
melted <- na.exclude(melted)
## Add marker numbers
marker_numbers <- data.frame(name = rownames(scan),
marker_number = 1:nrow(scan),
stringsAsFactors = FALSE)
melted <- inner_join(melted, marker_numbers)
## Recode sex to words
melted$sex_char <- ifelse(melted$sex == 1, "male", "female")
plot_scatter <- qplot(x = value,
geom = "jitter",
y = w212,
colour = factor(value),
data = melted) +
facet_wrap(~ factor(sex_char),
ncol = 1) +
xlab("Genotype") +
ylab("Body mass") +
formatting +
transition_manual(marker_number)
Combining the animations
And here is the final animation:

To put the pieces together, we use this magick trick (posted by Matt Crump). That is, animate the plots, one frame for each marker, and then use the R interface for ImageMagick to put them together and write them out.
gif_lod <- animate(plot_lod,
fps = 2,
width = 320,
height = 320,
nframes = sum(informative))
gif_scatter <- animate(plot_scatter,
fps = 2,
width = 320,
height = 320,
nframes = sum(informative))
## Magick trick from Matt Crump
mgif_lod <- image_read(gif_lod)
mgif_scatter <- image_read(gif_scatter)
new_gif <- image_append(c(mgif_lod[1], mgif_scatter[1]))
for(i in 2:sum(informative)){
combined <- image_append(c(mgif_lod[i], mgif_scatter[i]))
new_gif <- c(new_gif, combined)
}
image_write(new_gif, path = "out.gif", format = "gif")
Literature
Henriksen, Rie, et al. ”The domesticated brain: genetics of brain mass and brain structure in an avian species.” Scientific reports 6.1 (2016): 1-9.
