Last week’s post just happened to use MCMCglmm as an example of an R package that can get confused by tibble-style data frames. To make that example, I simulated some pedigree and trait data. Just for fun, let’s look at the simulation code, and use MCMCglmm and AnimalINLA to get heritability estimates.
First, here is some AlphaSimR code that creates a small random mating population, and collects trait and pedigree:
library(AlphaSimR)
## Founder population
FOUNDERPOP <- runMacs(nInd = 100,
nChr = 20,
inbred = FALSE,
species = "GENERIC")
## Simulation parameters
SIMPARAM <- SimParam$new(FOUNDERPOP)
SIMPARAM$addTraitA(nQtlPerChr = 100,
mean = 100,
var = 10)
SIMPARAM$setGender("yes_sys")
SIMPARAM$setVarE(h2 = 0.3)
## Random mating for 9 more generations
generations <- vector(mode = "list", length = 10)
generations[[1]] <- newPop(FOUNDERPOP,
simParam = SIMPARAM)
for (gen in 2:10) {
generations[[gen]] <- randCross(generations[[gen - 1]],
nCrosses = 10,
nProgeny = 10,
simParam = SIMPARAM)
}
## Put them all together
combined <- Reduce(c, generations)
## Extract phentoypes
pheno <- data.frame(animal = combined@id,
pheno = combined@pheno[,1])
## Extract pedigree
ped <- data.frame(id = combined@id,
dam = combined@mother,
sire =combined@father)
ped$dam[ped$dam == 0] <- NA
ped$sire[ped$sire == 0] <- NA
## Write out the files
write.csv(pheno,
file = "sim_pheno.csv",
row.names = FALSE,
quote = FALSE)
write.csv(ped,
file = "sim_ped.csv",
row.names = FALSE,
quote = FALSE)
In turn, we:
- Set up a founder population with a AlphaSimR’s generic livestock-like population history, and 20 chromosomes.
- Choose simulation parameters: we have an organism with separate sexes, a quantitative trait with an additive polygenic architecture, and we want an environmental variance to give us a heritability of 0.3.
- We store away the founders as the first generation, then run a loop to give us nine additional generations of random mating.
- Combine the resulting generations into one population.
- Extract phenotypes and pedigree into their own data frames.
- Optionally, save the latter data frames to files (for the last post).
Now that we have some data, we can fit a quantitative genetic pedigree model (”animal model”) to estimate genetic parameters. We’re going to try two methods to fit it: Markov Chain Monte Carlo and (the unfortunately named) Integrated Nested Laplace Approximation. MCMC explores the posterior distribution by sampling; I’m not sure where I heard it described as ”exploring a mountain by random teleportation”. INLA makes approximations to the posterior that can be integrated numerically; I guess it’s more like building a sculpture of the mountain.
First, a Gaussian animal model in MCMCglmm:
library(MCMCglmm)
## Gamma priors for variances
prior_gamma <- list(R = list(V = 1, nu = 1),
G = list(G1 = list(V = 1, nu = 1)))
## Fit the model
model_mcmc <- MCMCglmm(scaled ~ 1,
random = ~ animal,
family = "gaussian",
prior = prior_gamma,
pedigree = ped,
data = pheno,
nitt = 100000,
burnin = 10000,
thin = 10)
## Calculate heritability for heritability from variance components
h2_mcmc_object <- model_mcmc$VCV[, "animal"] /
(model_mcmc$VCV[, "animal"] + model_mcmc$VCV[, "units"])
## Summarise results from that posterior
h2_mcmc <- data.frame(mean = mean(h2_mcmc_object),
lower = quantile(h2_mcmc_object, 0.025),
upper = quantile(h2_mcmc_object, 0.975),
method = "MCMC",
stringsAsFactors = FALSE)
And here is a similar animal model in AnimalINLA:
library(AnimalINLA)
## Format pedigree to AnimalINLA's tastes
ped_inla <- ped
ped_inla$id <- as.numeric(ped_inla$id)
ped_inla$dam <- as.numeric(ped_inla$dam)
ped_inla$dam[is.na(ped_inla$dam)] <- 0
ped_inla$sire <- as.numeric(ped_inla$sire)
ped_inla$sire[is.na(ped_inla$sire)] <- 0
## Turn to relationship matrix
A_inv <- compute.Ainverse(ped_inla)
## Fit the model
model_inla <- animal.inla(response = scaled,
genetic = "animal",
Ainverse = A_inv,
type.data = "gaussian",
data = pheno,
verbose = TRUE)
## Pull out summaries from the model object
summary_inla <- summary(model_inla)
## Summarise results
h2_inla <- data.frame(mean = summary_inla$summary.hyperparam["Heritability", "mean"],
lower = summary_inla$summary.hyperparam["Heritability", "0.025quant"],
upper = summary_inla$summary.hyperparam["Heritability", "0.975quant"],
method = "INLA",
stringsAsFactors = FALSE)
If we wrap this all in a loop, we can see how the estimation methods do on replicate data (full script on GitHub). Here are estimates and intervals from ten replicates (black dots show the actual heritability in the first generation):
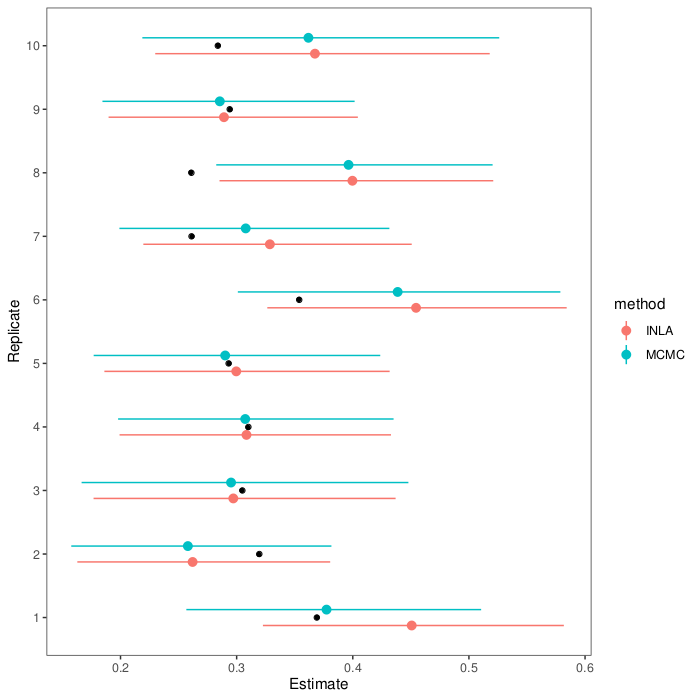
As you can see, the MCMC and INLA estimates agree pretty well and mostly hit the mark. In the one replicate dataset where they falter, they falter together.
