(This time it’s not just a Journal Club of One, because this post is based on a presentation given at the Hickey group journal club.)
The backstory goes like this: Polled cattle lack horns, and it would be safer and more convenient if more cattle were born polled. Unfortunately, not all breeds have a lot of polled cattle, and that means that breeding hornless cattle is difficult. Gene editing could help (see Bastiaansen et al. (2018) for a model).
In 2013, Tan et al. reported taking cells from horned cattle and editing them to carry the polled allele. In 2016, Carlson et al. cloned bulls based on a couple of these cell lines. The plan was to use the bulls, now grown, to breed polled cattle in Brazil (Molteni 2019). But a few weeks ago, FDA scientists (Norris et al 2019) posted a preprint that found inadvertent plasmid insertion in the bulls, using the public sequence data from 2016. Recombinetics, the company making the edited bulls, conceded that they’d missed the insertion.
”We weren’t looking for plasmid integrations,” says Tad Sonstegard, CEO of Recombinetics’ agriculture subsidiary, Acceligen, which was running the research with a Brazilian consulting partner. ”We should have.”
Oops.
For context: To gene edit a cell, one needs to bring both the editing machinery (proteins in the case of TALENS, the method used here; proteins and RNA in the case of CRISPR) and the template DNA into the cell. The template DNA is the DNA you want to put in instead of the piece that you’re changing. There are different ways to get the components into the cell. In this case, the template was delivered as part of a plasmid, which is a bacterially-derived circular DNA.
The idea is that the editing machinery should find a specific place in the genome (where the variant that causes polledness is located), make a cut in the DNA, and the cell, in its efforts to repair the cut, will incorporate the template. Crucially, it’s supposed to incorporate only the template, and not the rest of the plasmid. But in this case, the plasmid DNA snuck in too, and became part of the edited chromosome. Biological accidents happen.
How did they miss that, and how did the FDA team detect it? Both the 2016 and 2019 paper are short letters where a lot of the action is relegated to the supplementary materials. Here are pertinent excerpts from Carlson & al 2016:
A first PCR assay was performed using (btHP-F1: 5’- GAAGGCGGCACTATCTTGATGGAA; btHP-R2- 5’- GGCAGAGATGTTGGTCTTGGGTGT) … The PCR creates a 591 bp product for Pc compared to the 389 bp product from the horned allele.
Secondly, clones were analyzed by PCR using the flanking F1 and R1 primers (HP1748-F1- 5’- GGGCAAGTTGCTCAGCTGTTTTTG; HP1594_1748-R1- 5’-TCCGCATGGTTTAGCAGGATTCA) … The PCR creates a 1,748 bp product for Pc compared to the 1,546 bp product from the horned allele.
All PCR products were TOPO cloned and sequenced.
Thus, they checked that the animals were homozygotes for the polled allele (called ”Pc”) by amplifying two diagnostic regions and sequenced them to check the edit. This shows that the target DNA is there.
Then, they used whole-genome short read sequencing to check for off-target edits:
Samples were sequenced to an average 20X coverage on the Illumina HiSeq 2500 high output mode with paired end 125 bp reads were compared to the bovine reference sequence (UMD3.1).
Structural variations were called using CLC probabilistic variant detection tools, and those with >7 reads were further considered even though this coverage provides only a 27.5% probability of accurately detecting heterozygosity.
Upon indel calls for the original non-edited cell lines and 2 of the edited animals, we screened for de novo indels in edited animal RCI-001, which are not in the progenitor cell-line, 2120.
We then applied PROGNOS4 with reference bovine genome build UMD3.1 to compute all potential off-targets likely caused by the TALENs pair.
For all matching sequences computed, we extract their corresponding information for comparison with de novo indels of RCI-001 and RCI-002. BEDTools was adopted to find de novo indels within 20 bp distance of predicted potential targets for the edited animal.
Only our intended edit mapped to within 10 bp of any of the identified degenerate targets, revealing that our animals are free of off-target events and further supporting the high specificity of TALENs, particularly for this locus.
That means, they sequenced the animals’ genomes in short fragment, puzzled it together by aligning it to the cow reference genome, and looked for insertions and deletions in regions that look similar enough that they might also be targeted by their TALENs and cut. And because they didn’t find any insertions or deletions close to these potential off-target sites, they concluded that the edits were fine.
The problem is that short read sequencing is notoriously bad at detecting larger insertions and deletions, especially of sequences that are not in the reference genome. In this case, the plasmid is not normally part of a cattle genome, and thus not in the reference genome. That means that short reads deriving from the inserted plasmid sequence would probably not be aligned anywhere, but thrown away in the alignment process. The irony is that with short reads, the bigger something is, the harder it is to detect. If you want to see a plasmid insertion, you have to make special efforts to look for it.
Tan et al. (2013) were aware of the risk of plasmid insertion, though, at least when concerned with the plasmid delivering the TALEN. Here is a quote:
In addition, after finding that one pair of TALENs delivered as mRNA had similar activity as plasmid DNA (SI Appendix, Fig. S2), we chose to deliver TALENs as mRNA to eliminate the possible genomic integration of TALEN expression plasmids. (my emphasis)
As a sidenote, the variant calling method used to look for off-target effects (CLC Probabilistic variant detection) doesn’t even seem that well suited to the task. The manual for the software says:
The size of insertions and deletions that can be found depend on how the reads are mapped: Only indels that are spanned by reads will be detected. This means that the reads have to align both before and after the indel. In order to detect larger insertions and deletions, please use the InDels and Structural Variation tool instead.
The CLC InDels and Structural Variation tool looks at the unaligned (soft-clipped) ends of short sequence reads, which is one way to get at structural variation with short read sequences. However, it might not have worked either; structural variation calling is a hard task, and the tool does not seem to be built for this kind of task.
What did Norris & al (2019) do differently? They took the published sequence data and aligned it to a cattle reference genome with the plasmid sequence added. Then, they loaded the alignment into the trusty Integrative Genomics Viewer and manually looked for reads aligning to the plasmid and reads supporting junctions between plasmid, template DNA and genome. This bespoken analysis is targeted to find plasmid insertions. The FDA authors must have gone ”nope, we don’t buy this” and decided to look for the plasmid.
Here is what they claim happened (Fig 1): The template DNA is there, as evidenced by the PCR genotyping, but it inserted twice, with the rest of the plasmid in-between.
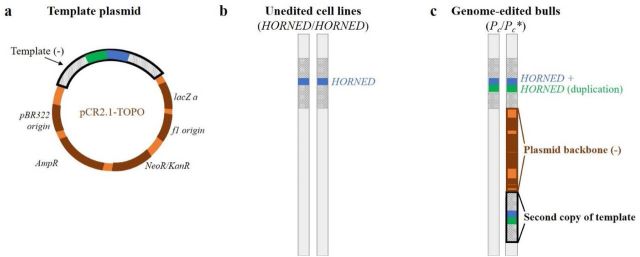
Here is the evidence (Supplementary figs 1 and 2): These are two annotated screenshots from IGV. The first shows alignments of reads from the calves and the unedited cell lines to the plasmid sequence. In the unedited cells, there are only stray reads, probably misplaced, but in the edited calves, ther are reads covering the plasmid throughout. Unless somehow else contaminated, this shows that the plasmid is somewhere in their genomes.
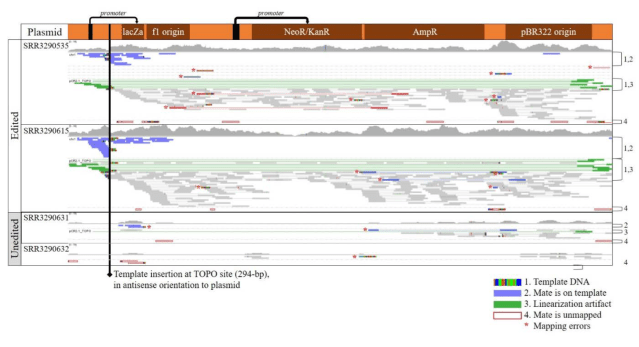
Where is it then? This second supplementary figure shows alignments to expected junctions: where template DNA and genome are supposed to join. The colourful letters are mismatches, showing where unexpected DNA shows up. This is the evidence for where the plasmid integrated and what kind of complex rearrangement of template, plasmid and genome happened at the cut site. This must have been found by looking at alignments, hypothesising an insertion, and looking for the junctions supporting it.
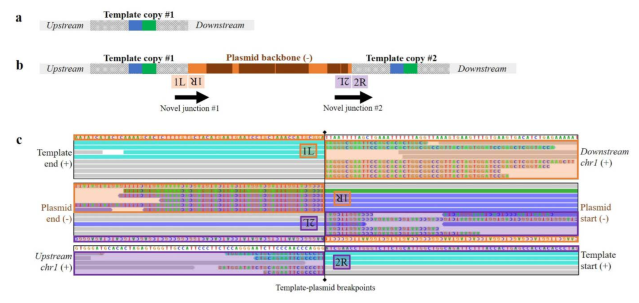
Why didn’t the PCR and targeted sequencing find this? As this third supplementary figure shows, the PCRs used could, theoretically, produce longer products including plasmid sequence. But they are way too long for regular PCR.
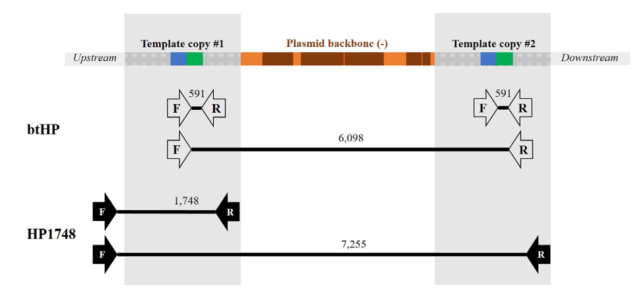
Looking at this picture, I wonder if there were a few attempts to make a primer pair that went from insert into the downstream sequence, that failed and got blamed on bad primer design or PCR conditions.
In summary, the 2019 preprint finds indirect evidence of the plasmid insertion by looking hard at short read alignments. Targeted sequencing or long read sequencing could give better evidence by observing he whole insertion. Recombinetics have acknowledged the problem, which makes me think that they’ve gone back to the DNA samples and checked.
Where does that leave us with quality control of gene editing? There are three kinds of problems to worry about:
- Off-target edits in similar places in other parts of the genome; this seems to be what people used to worry about the most, and what Carlson & al checked for
- Complex rearrangements around cut site (probably due to repeated cutting; this became a big concern after Kosicki & al (2018), and should apply both to on- and off-target cuts
- Insertion of plasmid or mutated target; this is what happened in here
The ways people check gene edits (targeted Sanger sequencing and short read sequencing) doesn’t detect any of them particularly well, at least not without bespoke analysis. Maybe the kind of analysis that Norris & al do could be automated to some extent, but currently, the state of the art seems to be to manually look closely at alignments. If I was reviewing the preprint, I would have liked it if the manuscript had given a fuller description of how they arrived at this picture, and exactly what the evidence for this particular complex rearrangement is. This is a bit hard to follow.
Finally, is this embarrassing? On the one hand, this is important stuff, plasmid integration is a known problem, so the original researchers probably should have looked harder for it. On the other hand, the cell lines were edited and the clones born before a lot of the discussion and research of off-target edits and on-target rearrangements that came out of CRISPR being widely applied, and when long read sequencing was a lot less common. Maybe it was easier to think that the sort read off-target analysis was enough then. In any case, we need a solid way to quality check edits.
Literature
Molteni M. (2019) Brazil’s plan for gene edited-cows got scrapped–here’s why. Wired.
Carlson DF, et al. (2016) Production of hornless dairy cattle from genome-edited cell lines. Nature Biotechnology.
Norris AL, et al. (2019) Template plasmid integration in germline genome-edited cattle. BioRxiv.
Tan W, et al. (2013) Efficient nonmeiotic allele introgression in livestock using custom endonucleases. Proceedings of the National Academy of Sciences.
Bastiaansen JWM, et al. (2018) The impact of genome editing on the introduction of monogenic traits in livestock. Genetics Selection Evolution.
Kosicki M, Tomberg K & Bradley A. (2018) Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nature Biotechnology.
