So, a couple of weeks ago I tweeted from the @sweden account. This is a short recap of some things that were said, and a few links that I promised people. Overall I think it went pretty well. I didn’t tweet as much as some other curators, but much much more than I usually do. This also meant I did spend my lunch and coffee breaks looking at my phone. My tweets are collected here, if for some reason you’d care to read them.
Of course, tweeting from a rotating curation account is very different from the way I normally use Twitter. First, I read much more than I write. One of the main purposes of Twitter, for me, is to get a steady stream of links to read. That doesn’t really work on an account that follows much more and entirely different people. A lot of what I wrote was prepared monologue, but I don’t think that’s necessarily a bad thing. I follow a lot of people on Twitter for their monologues. Also, thankfully, a lot of people asked me questions! Another thing that struck me is that so few people were unpleasant. There were a few extreme right folks who wanted me to retweet their racist tweets, but only a few. Then, a few felt the need to tell me that I’m utterly boring, which is fine. Someone lamented the fact that all curators are uneducated about the proper use of Twitter (it’s probably to build your personal brand or something). Also, a certain Swedish celebrity got put on ignore so I wouldn’t have to see him tagging each tweet with ”@sweden”. But that was pretty much all.
I talked quite a bit about my research. I spent more or less a full day on the chicken comb as a sexual ornament and genetics of comb mass. We discussed domestication as an evolutionary process, tonic immobility, and how to measure gene expression for eQTL mapping. I also wrote about Kauai feral chickens … And what I actually do in a day nowadays, that is: writing R code.
For anyone who wonders what my work looks like today, I took a picture. http://t.co/dfmw07iPXZ—
@sweden / Anya (@sweden) April 13, 2015
I got a question about what to say to your creationist friend. I think this depends on what the creationist friend believes and what their objections to evolution are. Unfortunately, I don’t think there is a simple knock-down argument against all forms of creationism, except that evolution works really well and has a lot of evidence going for it. I certainly don’t think it will do to rely on methodological naturalism and say that ”creation would be a supernatural event and outside the scope of science”. First, because I don’t think that is how science works. Say if unicorns, miraculous healing, and species popping into existence without relation to other species were actually part of the world, wouldn’t we want to study that? Second, that will never convince anyone, except of the irrelevance of science to their worldview.
But I think there are a handful of things that creationists often take issue with. First, some don’t believe in sequence variants creating new functions. This is often described with slogans about information, and how it cannot be created by random mutation. I don’t think ”sequence information” is a particularly useful concept, and would much prefer to talk about function and adaptation. That is what is important, after all, organisms acquiring new adaptations. It turns out, new functions arising can be observed, particularly in microorganisms. Some really fun and well-studied example occur in the Long Term Evolution Experiment; see Richard Lenski’s blog which has explanatory posts and links to papers.
Second, the formation of species come up a lot in these discussions. This is a bit tricky, because it’s not always clear what constitutes different species. The definition most people have heard is probably that individuals belong to different species if they cannot have fertile offspring. But just think of asexually reproducing organisms. There, individuals belong to different species if they’re sufficiently different. So we already have what is needed to understand the formation of species in the evolution of new functions. When it comes to sexually reproducing organisms, there are examples of the evolution of reproductive isolation — cases where it seems to be ongoing or to have happened recently. (See for instance this paper on hybrid incompatibility in Mimulus guttatus; I have blogged about it, but only in Swedish)
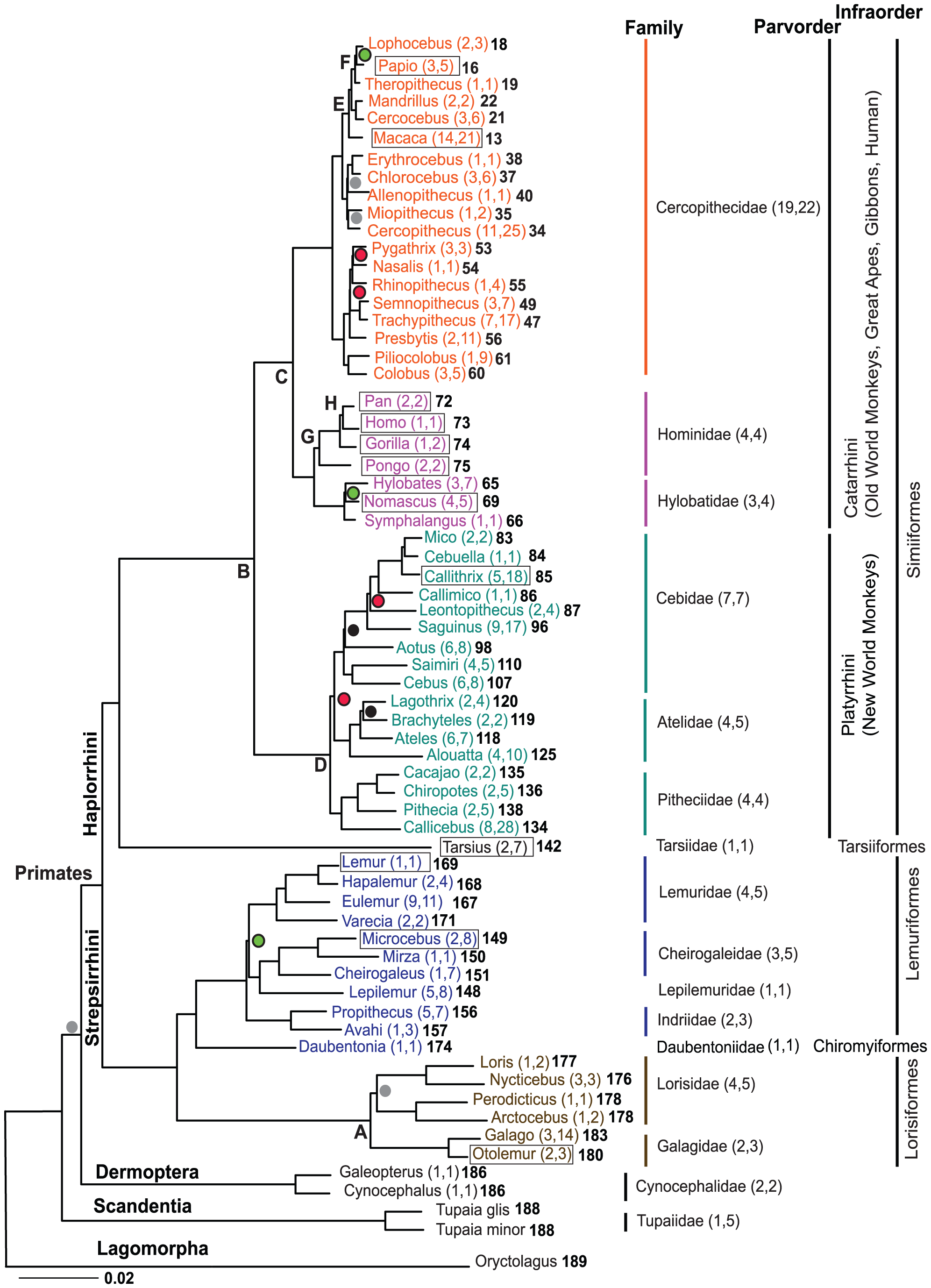
Third, there is the question of relatedness between species. In particular, some creationists really hate the idea that humans are apes. I think it is important to emphasize a couple of things that evolution does not say about humans and other apes. By the way, this isn’t just confusing for creationists, but for everyone. Evolution does not mean that humans descend from extant apes. Look at this phylogenetic tree from Perelman & al 2011. This is just like a family tree, but of populations: we see how chimps and humans have a recent common ancestor population. This is different than claiming that we would descend from extant chimps. Of course, chimps have also changed since the common ancestor, although not in the same ways as humans. (Again, I’ve written about this before in Swedish.)
Speaking of unicorns, I of course celebrated unicorn Friday:
#unicornfriday http://t.co/SSQrBuUi9j—
@sweden / Patrik (@sweden) April 17, 2015
Someone asked whether you can keep fruit flies for amateur genetics at home. That should be quite possible, and I don’t see any real problems with it either. The fruit fly community has really strong culture of classical genetics with crosses and stocks. I don’t know if stock centres would deliver to private customers, but I don’t see why they wouldn’t — except for transgenic flies. It turned out, however, that transgenic flies was actually what the person asking was after. And of course, I can’t recommend that. I must say, I have mixed feelings about do-it-yourself biotechnology. On the one hand, some home molecular biology should be possible and rewarding. On the other hand, a lot of things routinely used in molecular labs are actually really dangerous if misused, and not just for the user. For example, when making any type of construct in transgenic bacteria, antibiotics and antibiotic resistance genes are the standard screening markers. They are used to pick out the bacteria that have incorporated the piece of DNA you care about. This is not the kind of stuff you want to use without proper containment. So, in the fly example, you would not only have to handle the flies, but also transgenic antibiotic resistant bacteria safely and legally. Then again, a lot of the genetics I care about does not involve any of that, and could very well be done in a basement.
The @sweden account caught me under a teaching week; otherwise, all of my photos would’ve been my computer, my pen and my coffee mug. Now I got to walk the followers through agarose gel electrophoresis and a little transformation of bacteria:
We put the electric field on, and DNA, negatively charged, travels through the gel towards the positive pole. http://t.co/fE3w5A7Vle—
@sweden / Patrik (@sweden) April 14, 2015
Guess the process! http://t.co/D4gcogBe0p—
@sweden / Patrik (@sweden) April 16, 2015
And, finally, Swedish spring:
The evening walk. http://t.co/ncPQf50aiN—
@sweden / Patrik (@sweden) April 18, 2015