In September, I went to the 20th Evolutionary Biology Meeting in Marseille. This is a very nice little meeting. I listened to a lot of talks, had some very good conversations, met some people, and presented our effort to map domestication traits in the chicken with quantitative trait locus mapping and gene expression (Johnsson & al 2015, 2016, and some unpublished stuff).
Time for a little conference report. Late, but this time less than a year from the actual conference. Here are some of my highlights:
Richard Cordaux on pill bugs, Wolbachia and sex manipulation — I did not know that Wolbachia, the intracellular parasite superstar of arthropods, had feminization of hosts in its repertoire (Cordaux & al 2004). Not only that, but in some populations of pill bugs, a large chunk of the genome of the feminizing Wolbachia has inserted into the pill bug genome, thus forming a new W chromosome (Leclercq & al 2016, published since the conference). He also told me how this is an example of the importance of preserving genetic resources — the lines of pill bugs have been maintained for a long time, and now they’re able to return to them with genomics tools and continue old lines of research. I think that is seriously cool.
Olaya Rendueles Garcia on positive frequency-dependent selection maintaining diversity in social bacterium Myxococcus xanthus (Rendueles, Amherd & Velicer 2015) — In my opinion, this was the best talk of the conference. It had everything: an interesting phenomenon, a compelling study system, good visuals and presentation. In short: M. xanthus of the same genotype tend to cooperate, inhabit their own little turfs in the soil, and exclude other genotypes. So it seems positive frequency-dependent selection maintains diversity in this case — diversity across patches, that is.
A very nice thing about this kind of meetings is that one gets a look into the amazing diversity of organisms. Or as someone put it: the complete and utter mess. In this department, I was particularly struck by … Sally Leys — sponges; Marie-Claude Marsolier-Kergoat — bison; Richard Dorrell — stramenopile chloroplasts.
I am by no means a transposable elements person. In fact, one might believe I was actively avoiding transposable elements by my choice of study species. But transposable elements are really quite interesting, and seem quite important to genome evolution, both to neutrally evolving and occasionally adaptive sequences. This meeting had a good transposon session, with several interesting talks.
Anton Crombach presented models the gap gene network in Drosophila melanogaster and Megaselia abdita, with some evolutionary perspectives (Crombach & al 2016). A couple of years ago, Marjoram, Zubair & Nuzhdin used the gap gene network as their example model to illustrate the suggestion to combine systems biology models with genetic mapping. I very much doubt (though I may be wrong; it happens a lot) that there is much meaningful variation within populations in the gap gene network. A between-species analysis seems much more fruitful, and leads to the interesting result where the outcome, in terms of gap gene expression along the embryo, is pretty similar but the way that the system gets there is quite different.
If you’ve had a beer with me and talked about the future of quantitative genetics, you’re pretty likely to have heard me talk about how in the bright future, we will not just map variation in phenotypes, but in the parameters of dynamical models. (I also think that the mapping will take place through fully Bayesian hierarchical models where the same posterior can be variously summarized for doing genomic prediction or for mapping the major quantitative trait genes, interactions etc. Of course, setting up and running whole-genome long read sequencing will be as convenient and cheap as an overnight PCR. And generally, there will be pie in the sky etc.) At any rate, what Anton Crombach showed was an example of combining systems biology modelling with variation (between clades). I thought it was exciting.
It was fun to hear Didier Raoult, one of the discoverers of giant viruses, speak. He was somewhat of a quotation machine.
”One of the major problems in biology is that people believe what they’ve learned.”
(About viruses being alive or not) ”People ask: are they alive, are they alive? I don’t care, and they don’t care either”
Very entertaining, and quite fascinating stuff about giant viruses.
If there are any readers out there who worry about social media ruining science by spilling the beans about unpublished results presented at meetings, do not worry. There were a few more cool unpublished things. Conference participants, you probably don’t know who you are, but I eagerly await your papers.
I think this will be the last evolution-themed conference for me in a while. The EBM definitely has a different range of themes than the others I’ve been to: ESEB, or rather: the subset of ESEB I see choosing my adventure through the multiple-session programme, and the Swedish evolution meetings. There was more molecular evolution, more microorganisms and even some orgin of life research.







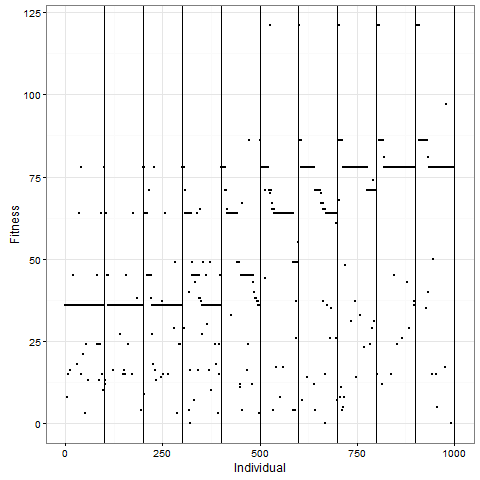
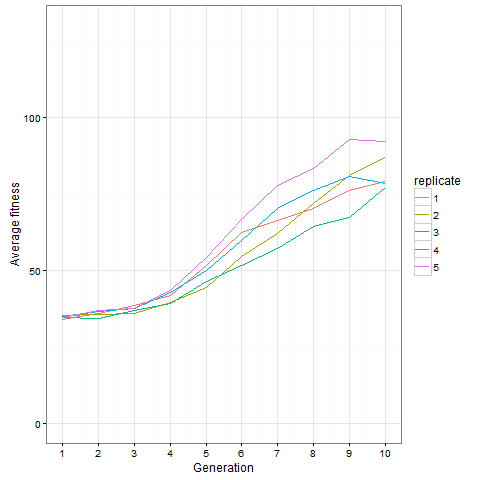
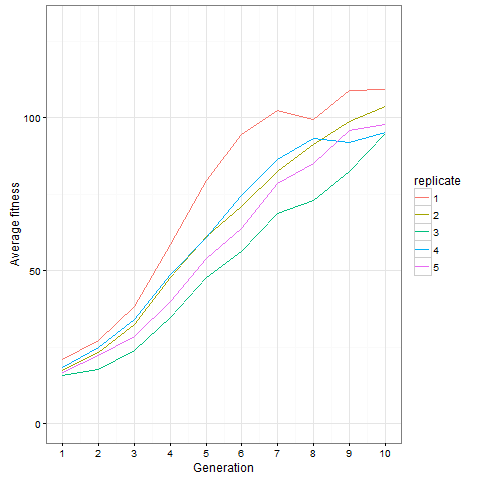
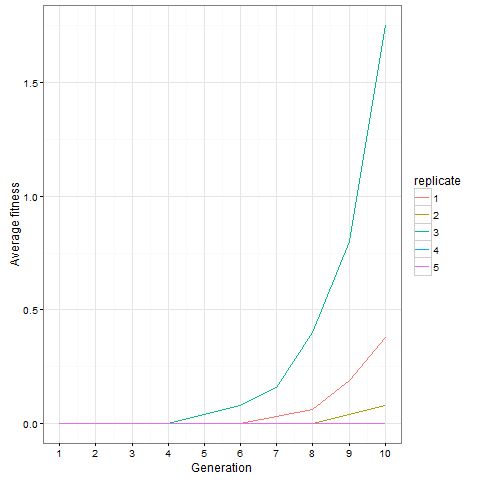
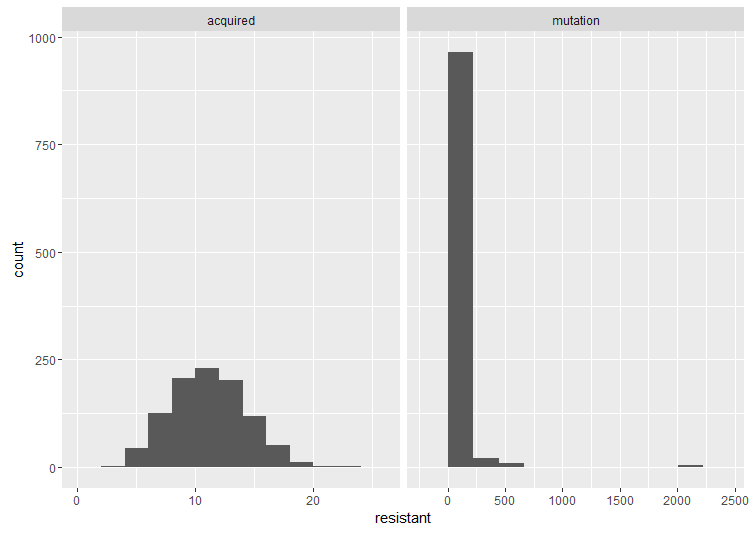
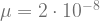 , which is quite close to their estimated value, and that the mutation can’t reverse. We also assume that the bacteria grow by doubling each generation up to 30 generations. We start a culture from a single susceptible bacterium, and let it grow for a number of generations before the phage is added. (We’re going to use discrete generations, while Luria & Delbrück use a continuous function.) Then:
, which is quite close to their estimated value, and that the mutation can’t reverse. We also assume that the bacteria grow by doubling each generation up to 30 generations. We start a culture from a single susceptible bacterium, and let it grow for a number of generations before the phage is added. (We’re going to use discrete generations, while Luria & Delbrück use a continuous function.) Then:

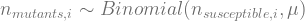 .
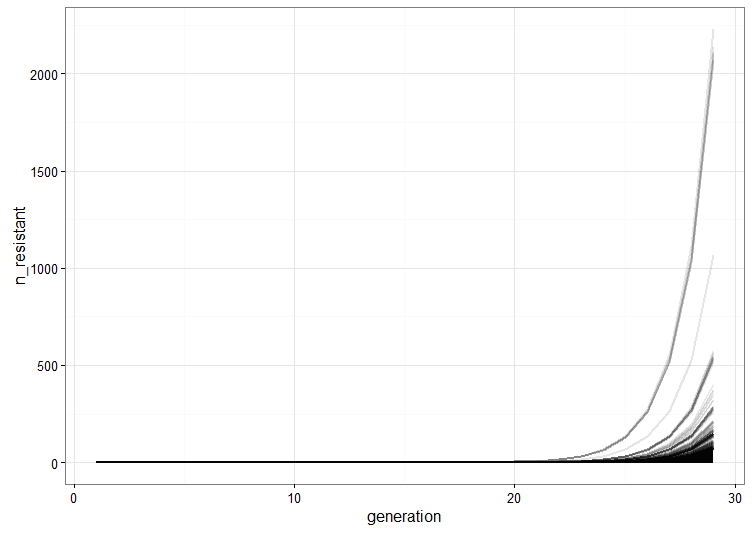
.
 .
.