How do allele frequencies change in response to selection? Answers to that question include ”it depends”, ”we don’t know”, ”sometimes a lot, sometimes a little”, and ”according to a nonlinear differential equation that actually doesn’t look too horrendous if you squint a little”. Let’s look at a model of the polygenic adaptation of an infinitely large population under stabilising selection after a shift in optimum. This model has been developed by different researchers over the years (reviewed in Jain & Stephan 2017).
Here is the big equation for allele frequency change at one locus:

That wasn’t so bad, was it? These are the symbols:
- the subscript i indexes the loci,
is the change in allele frequency per time,
is the effect of the locus on the trait (twice the effect of the positive allele to be precise),
is the frequency of the positive allele,
the frequency of the negative allele,
is the strength of selection,
is the phenotypic mean of the population; it just depends on the effects and allele frequencies
is the mutation rate.
This breaks down into three terms that we will look at in order.
The directional selection term

is the term that describes change due to directional selection.
Apart from the allele frequencies, it depends on the strength of directional selection 
It is negative because it describes the change in the allele with a positive effect on the trait, so if the mean phenotype is above the optimum, we would expect the allele frequency to decrease, and indeed: when
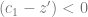
this term becomes negative.
If you neglect the other two terms and keep this one, you get Jain & Stephan's "directional selection model", which describes behaviour of allele frequencies in the early phase before the population has gotten close to the new optimum. This approximation does much of the heavy lifting in their analysis.
The stabilising selection term
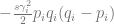
is the term that describes change due to stabilising selection. Apart from allele frequencies, it depends on the square of the effect of the locus on the trait. That means that, regardless of the sign of the effect, it penalises large changes. This appears to make sense, because stabilising selection strives to preserve traits at the optimum. The cubic influence of allele frequency is, frankly, not intuitive to me.
The mutation term
Finally,
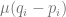
is the term that describes change due to new mutations. It depends on the allele frequencies, i.e. how of the alleles there are around that can mutate into the other alleles, and the mutation rate. To me, this is the one term one could sit down and write down, without much head-scratching.
Walking in allele frequency space
Jain & Stephan (2017) show a couple of examples of allele frequency change after the optimum shift. Let us try to draw similar figures. (Jain & Stephan don’t give the exact parameters for their figures, they just show one case with effects below their threshold value and one with effects above.)
First, here is the above equation in R code:
pheno_mean <- function(p, gamma) {
sum(gamma * (2 * p - 1))
}
allele_frequency_change <- function(s, gamma, p, z_prime, mu) {
-s * gamma * p * (1 - p) * (pheno_mean(p, gamma) - z_prime) +
- s * gamma^2 * 0.5 * p * (1 - p) * (1 - p - p) +
mu * (1 - p - p)
}
With this (and some extra packaging; code on Github), we can now plot allele frequency trajectories such as this one, which starts at some arbitrary point and approaches an optimum:

Animation of alleles at two loci approaching an equilibrium. Here, we have two loci with starting frequencies 0.2 and 0.1 and effect size 1 and 0.01, and the optimum is at 0. The mutation rate is 10-4 and the strength of selection is 1. Animation made with gganimate.
Resting in allele frequency space
The model describes a shift from one optimum to another, so we want want to start at equilibrium. Therefore, we need to know what the allele frequencies are at equilibrium, so we solve for 0 allele frequency change in the above equation. The first term will be zero, because
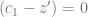
when the mean phenotype is at the optimum. So, we can throw away that term, and factor the rest equation into:

Therefore, one root is 

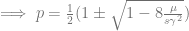
These points correspond to mutation–selection balance with one or the other allele closer to being lost. Jain & Stephan (2017) show a figure of the three equilibria that looks like a semicircle (from the quadratic equation, presumably) attached to a horizontal line at 0.5 (their Figure 1). Given this information, we can start our loci out at equilibrium frequencies. Before we set them off, we need to attend to the effect size.
How big is a big effect? Hur långt är ett snöre?
In this model, there are big and small effects with qualitatively different behaviours. The cutoff is at:

If we look again at the roots to the quadratic equation above, they can only exist as real roots if

because otherwise the expression inside the square root will be negative. This inequality can be rearranged into:
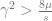
This means that if the effect of a locus is smaller than the threshold value, there is only one equilibrium point, and that is at 0.5. It also affects the way the allele frequency changes. Let us look at two two-locus cases, one where the effects are below this threshold and one where they are above it.
threshold <- function(mu, s) sqrt(8 * mu / s) threshold(1e-4, 1)
[1] 0.02828427
With mutation rate of 10-4 and strength of selection of 1, the cutoff is about 0.028. Let our ”big” loci have effect sizes of 0.05 and our small loci have effect sizes of 0.01, then. Now, we are ready to shift the optimum.
The small loci will start at an equilibrium frequency of 0.5. We start the large loci at two different equilibrium points, where one positive allele is frequent and the other positive allele is rare:
get_equilibrium_frequencies <- function(mu, s, gamma) {
c(0.5,
0.5 * (1 + sqrt(1 - 8 * mu / (s * gamma^2))),
0.5 * (1 - sqrt(1 - 8 * mu / (s * gamma^2))))
}
(eq0.05 <- get_equilibrium_frequencies(1e-4, 1, 0.05))
[1] 0.50000000 0.91231056 0.08768944
get_equlibrium_frequencies(1e-4, 1, 0.01)
[1] 0.5 NaN NaN
Look at them go!
These animations show the same qualitative behaviour as Jain & Stephan illustrate in their Figure 2. With small effects, there is gradual allele frequency change at both loci:
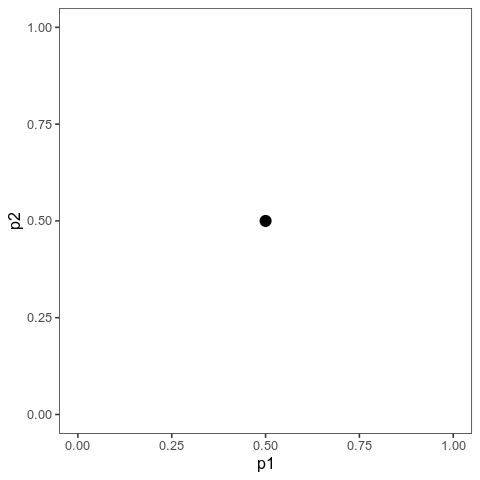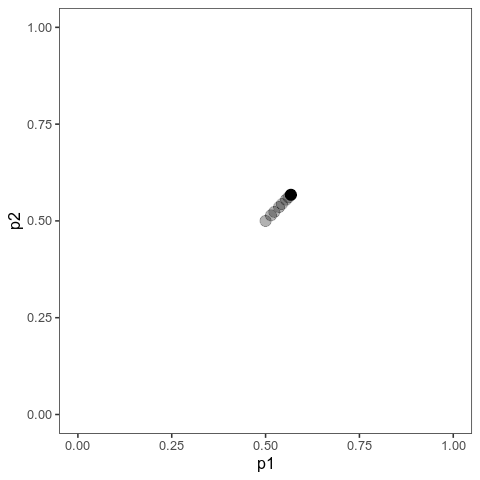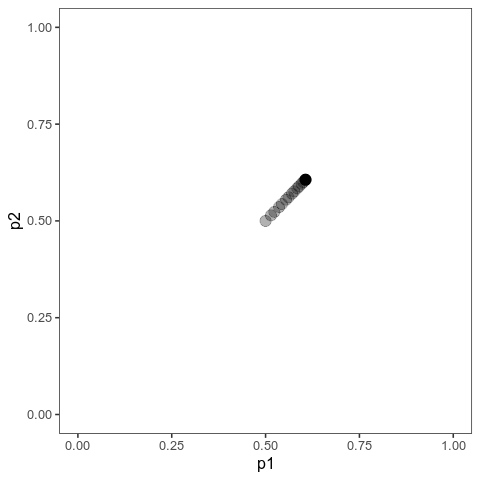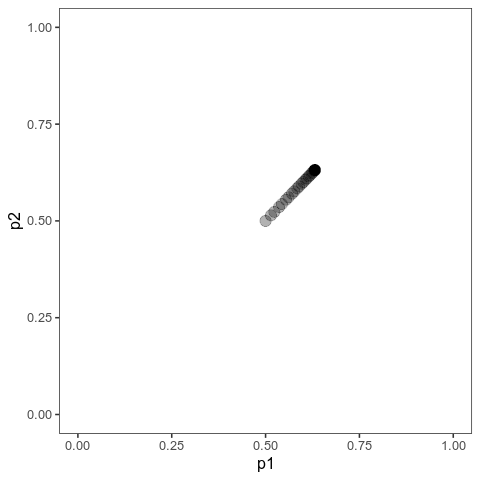
However, with large effects, one of the loci (the one on the vertical axis) dramatically changes in allele frequency, that is it’s experiencing a selective sweep, while the other one barely changes at all. And the model will show similar behaviour when the trait is properly polygenic, with many loci, as long as effects are large compared to the (scaled) mutation rate.

Here, I ran 10,000 time steps; if we look at the phenotypic means, we can see that they still haven’t arrived at the optimum at the end of that time. The mean with large effects is at 0.089 (new optimum of 0.1), and the mean with small effects is 0.0063 (new optimum: 0.02).
Let’s end here for today. Maybe another time, we can return how this model applies to actually polygenic architectures, that is, with more than two loci. The code for all the figures is on Github.
Literature
Jain, K., & Stephan, W. (2017). Modes of rapid polygenic adaptation. Molecular biology and evolution, 34(12), 3169-3175.





