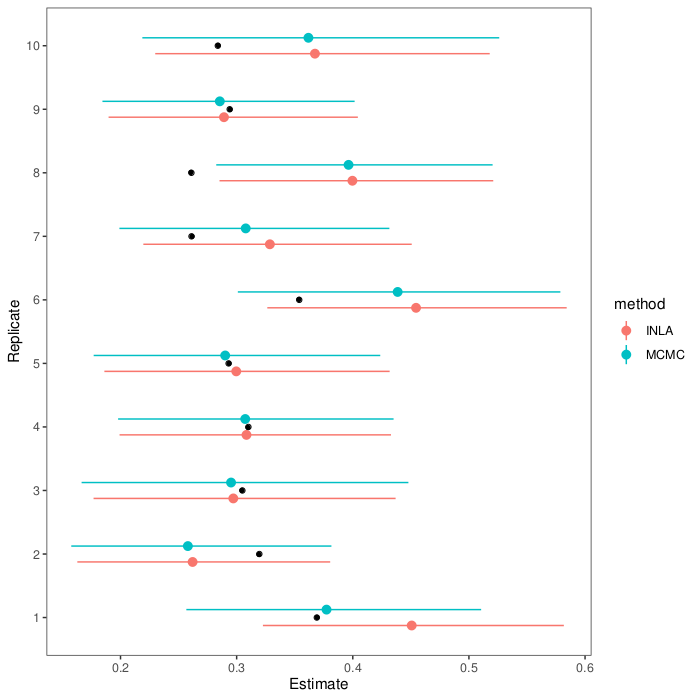
Tidy Tuesday is a fun weekly activity where a lot of R enthusiasts make different visualisations, and possibly modelling, of the same dataset. You can read more about it at their Github page. I participated for three weeks, and here is a recap. I will show excerpts of the code, but you can read the whole thing by clicking through to Github.
2019-10-22 Horror films
Data: https://github.com/rfordatascience/tidytuesday/tree/master/data/2019/2019-10-22
My code: https://github.com/mrtnj/rstuff/blob/master/tidytuesday/horror_movies.R
In time for Halloween, we got a dataset with horror film data from IMDB. (Yes, I will be mixing the terms ”film” and ”movie” wildly.)
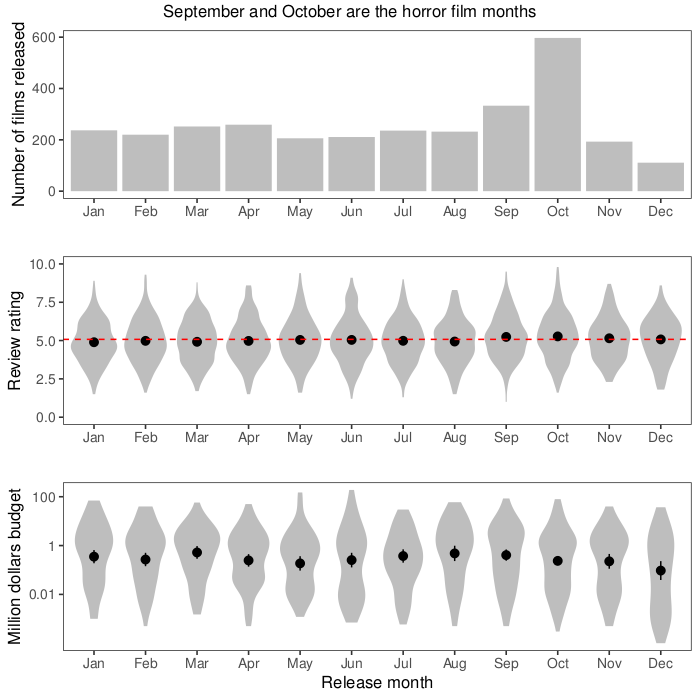
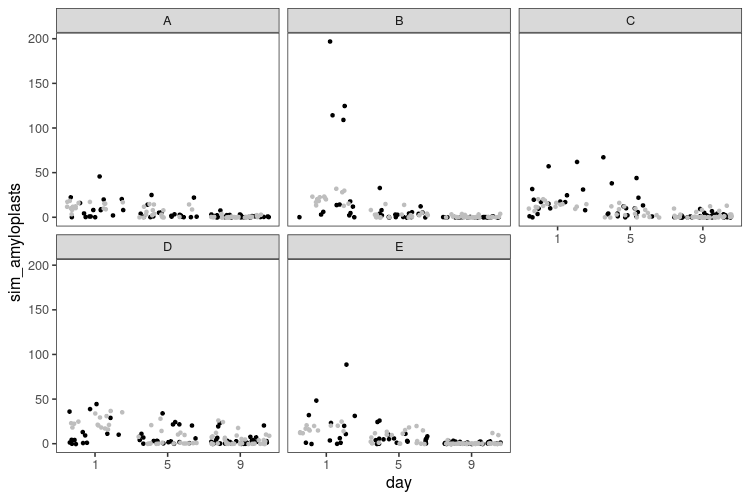
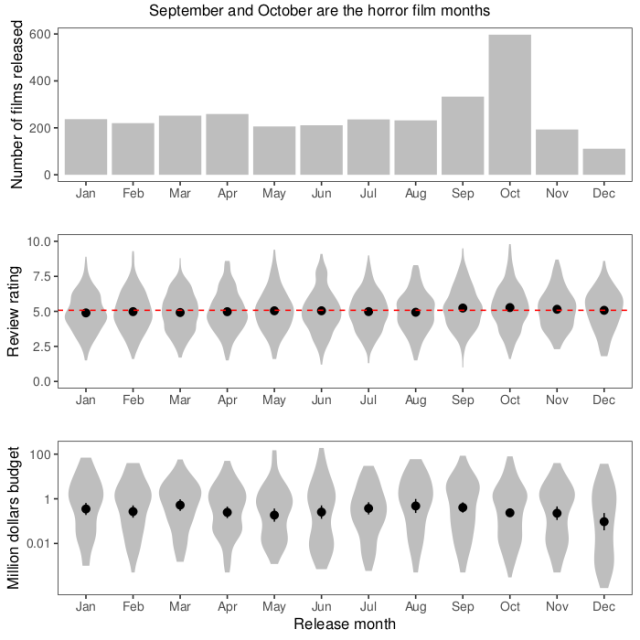
The first week, I started with making a pretty boring plot, the way I’d normally plot things (white background, small multiples, you know the drill). I wanted to look at distribution over the year, so I plotted what month films are released and the distribution of review scores and budgets each month. After thinking about it for a while, I thought a logarithmic scale would make sense for budgets, that span a huge range. Also, after realising that the budget column actually didn’t contain dollars, but a mix of currencies, I decided not to try to convert, but use only the US dollar budgets.
I don’t often run into dates, to using the date functions from readr and lubridate was new to me, as was the built-in vector month.abb:
library(dplyr)
library(egg)
library(ggplot2)
library(ggimage)
library(lubridate)
library(readr)
library(stringr)
movies <- read_csv("horror_movies.csv")
## Parse dates
movies$release_parsed <- parse_date(movies$release_date,
format = "%d-%b-%y",
locale = locale("en"))
movies$release_year <- ifelse(is.na(movies$release_parsed),
movies$release_date,
year(movies$release_parsed))
movies$release_month <- month.abb[month(movies$release_parsed)]
Here, we parse the release data, and extract the release year, treating films that only have a release year separately.
I also put in means with confidence intervals, like so, and a line for the mean review rating:
model <- lm(review_rating ~ release_month, movies)
fit <- data.frame(release_month = month.abb,
predict(model,
newdata = data.frame(release_month = month.abb),
interval = "confidence"),
stringsAsFactors = FALSE)
grand_mean_rating <- mean(movies$review_rating,
na.rm = TRUE)
As an example of the plotting code, here is the middle panel for ratings. As usual with ggplot2, we layer geometries on top of each other (here: violin plots, points with range bars, and a horizontal line, followed by a lot of formatting.
plot_rating <- ggplot() +
geom_violin(aes(x = release_month,
y = review_rating),
fill = "grey",
colour = NA,
data = movies) +
scale_x_discrete(limits = month.abb) +
geom_pointrange(aes(x = release_month,
y = fit,
ymax = upr,
ymin = lwr),
data = fit) +
geom_hline(yintercept = grand_mean_rating,
linetype = 2,
colour = "red") +
ylim(0, 10) +
theme_bw(base_size = 12) +
theme(panel.grid = element_blank()) +
xlab("") +
ylab("Review rating")
There is similar code for the other two panels. Finally, I used ggarrange from the egg package to put everything together. In summary, most horror films are released in October, probably around Halloween. The review ratings of films released in this horror season are also a tiny bit higher than during the rest of the year, but there is not much of a difference in the budgets.
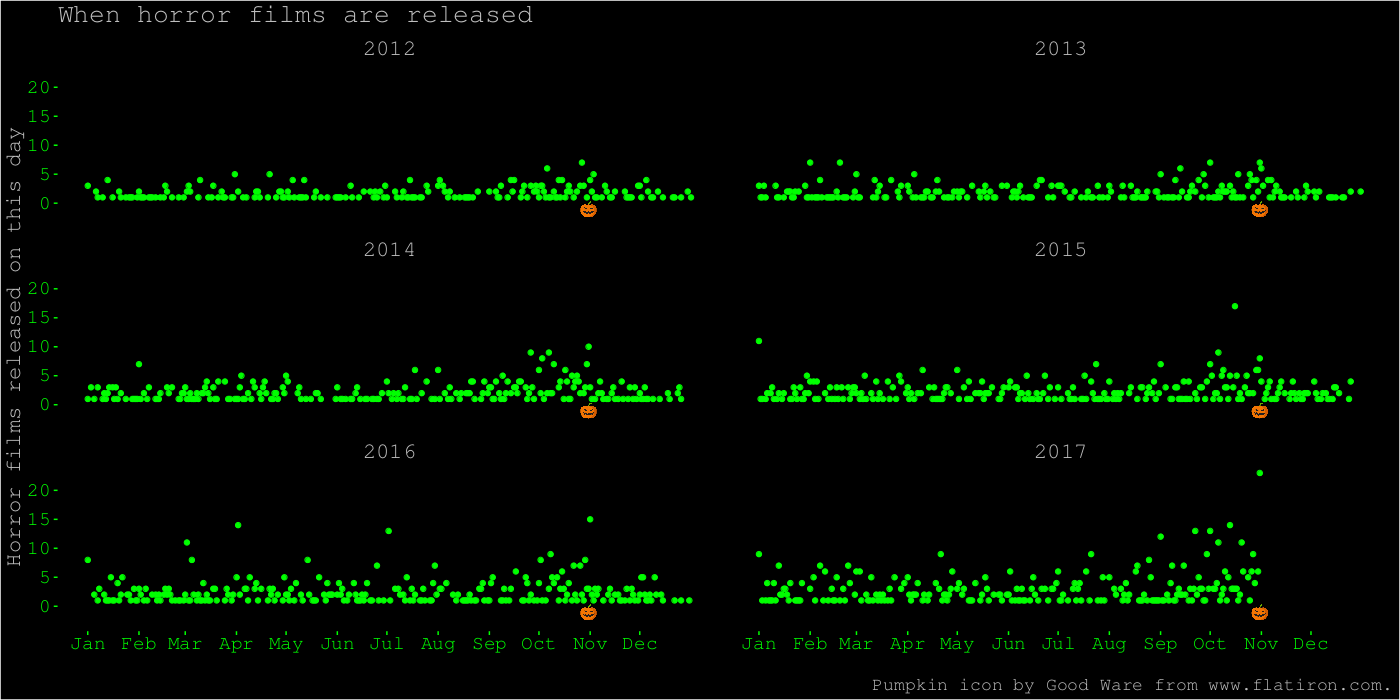
After that, and after seeing some of the fun horror-themed graphs other people made, I decided to make something more colourful. Here is a plot on the same theme, showing each day and year separately, an appropriately horrendous colour scheme, and a pumpkin icon to indicate the date of Halloween. I like this plot better because it shows more of the data. It shows the increase at Halloween. We also see some spikes at other dates, like 1 January of some years. It also shows how the dataset ends at Halloween 2017.
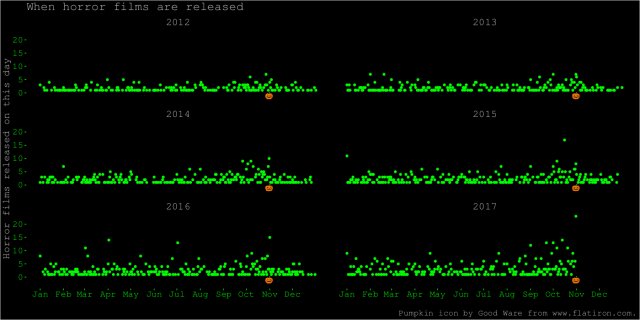
The code for this plot is mostly a lot of theme formatting. The ggplot2 theme function takes a lot of arguments I’ve never used before.
movies$yday <- yday(movies$release_parsed)
daycount <- summarise(group_by(movies, yday, release_year), n = n())
First, we turn dates into days of the year, and count the number of film releases.
halloween <- yday("2019-10-31")
pumpkin_data <- data.frame(x = halloween,
y = -1,
image = "pumpkin.png",
stringsAsFactors = FALSE)
Then, we set up the date of Halloween and a data frame for the pumpkin icon. We’re going to use geom_image from the ggimage package to add this icon to each subplot.
breaks <- yday(paste("2019-", 1:12, "-01", sep = ""))
plot_year <- ggplot() +
geom_point(aes(x = yday,
y = n),
colour = "green",
data = na.exclude(dc)) +
geom_image(aes(x = x,
y = y,
image = image),
data = pumpkin_data) +
facet_wrap(~ release_year,
ncol = 2) +
scale_x_continuous(breaks = breaks,
labels = month.abb) +
ylim(-3, NA) +
labs(caption = "Pumpkin icon by Good Ware from www.flatiron.com.") +
theme(panel.grid = element_blank(),
strip.background = element_blank(),
text = element_text(family = "mono",
colour = "grey",
size = 16),
axis.text = element_text(family = "mono",
colour = "green",
size = 14),
axis.ticks = element_line(colour = "green"),
strip.text = element_text(family = "mono",
colour = "grey",
size = 16),
plot.background = element_rect(fill = "black"),
panel.background = element_rect(fill = "black")) +
xlab("") +
ylab("Horror films released on this day") +
ggtitle("When horror films are released")
A lot of other people made graphs that highlight the increase in horror film releases around Halloween in different ways. Here are some that I like:
https://twitter.com/veerlevanson/status/1187770362588811266
And, looking deeper, there is a pattern within months too:
Finally, I also like this plot, that makes a case for a U-shaped relationship between budget and rating:
And for contrast, another that makes a different case with the same data:
This seems to be a recurrent theme when it comes to interpretation and quantitative analysis in the Tidy Tuesday datasets. People make different modeling choices, or visualisation choices (which are modeling choices) about what to lump together, what to separate into bins, how to transform the data, and how to show uncertainty. In some cases, as with the pattern of film releases around Halloween, they all find similar results. In some other cases, they don’t.
2019-10-28 NYC Squirrel Census
Data: https://github.com/rfordatascience/tidytuesday/tree/master/data/2019/2019-10-29
My code: https://github.com/mrtnj/rstuff/blob/master/tidytuesday/nyc_squirrels.R
This week, the data was about the location and activities of squirrels in New York central park on certain times. I had this vision of an animated map of squirrel locations. I ended up with an animation, but no map. The colour of the squirrel icon shows the main fur colour of the squirrels (grey, black, cinnamon), and the size shows adults and juveniles.

I had never used gganimate before (only animation, as in this post about the Game of Life), but I had seen Thomas Lin Pedersen tweet about it, and I wanted to try.
library(dplyr)
library(gganimate)
library(ggimage)
library(ggplot2)
library(readr)
squirrels <- read_csv("nyc_squirrels.csv")
## Parse the date
squirrels$date_parsed <- parse_date(as.character(squirrels$date), format = "%m%d%Y")
## Give each observation a unique ID (to use as group in the
## animation, so as to not have points turn into one another but fade
## instead.
squirrels$key <- 1:nrow(squirrels)
## Associate the different squirrel colours with the filenames of
## icons in different colours (manually filled with GIMP).
squirrels$image <- "squirrel.png"
squirrels$image[squirrels$primary_fur_color == "Cinnamon"] <- "squirrel_cinnamon.png"
squirrels$image[squirrels$primary_fur_color == "Gray"] <- "squirrel_grey.png"
squirrels$image[is.na(squirrels$primary_fur_colour)] <- NA
Again, we need to parse the date. We already have latitude and longitude. We need a unique identifier for each observation, to tell gganimate that we want each squirrel to be in its own group. Then, we associate squirrel colours with three different files with a squirrel icon in different colours.
First, we make two image scatterplot layers, setting the sizes of adults and juveniles manually. The colour is deal with by mapping the image column containing the file names to the image aesthetic. We add some formatting, and then, the transition_states layer, which is where the graph turns from still and boring to magical moving pictures. This will animate a series of discrete ”states”, which here consist of the date pasted together with the shift (AM or PM squirrel observation shift). The special ”{closest_state}” variable in the title string puts this state name as plot title.
plot_colour <- ggplot() +
geom_image(aes(y = long, x = lat, image = image, group = key),
size = 0.04,
data = filter(squirrels, age == "Adult")) +
geom_image(aes(y = long, x = lat, image = image, group = key),
size = 0.03,
data = filter(squirrels, age == "Juvenile")) +
theme_bw(base_size = 16) +
theme(panel.grid = element_blank()) +
xlab("Latitude") +
ylab("Longitude") +
labs(title = "{closest_state}",
caption = "Data from NYC Squirrel Census. Squirrel icon made by Freepik from www.flatiron.com.") +
transition_states(paste(date_parsed, shift),
state_length = 2,
transition_length = 1)
## Render it and write to file
animate(plot_colour,
fps = 10,
nframes = 400,
end_pause = 20,
rewind = FALSE,
width = 1000,
height = 1000)
I was faffing around unsuccessfully with different map packages to try to find something of Central Park. It seems ggmaps is the way to go. Several other participants made nice maps:
However, I think this was my favourite:
https://github.com/ryantimpe/TidyTuesday/blob/master/2019w44/2019w44.R
The original Squirrel Census Report seems to be amazing object, too, with a beautiful map.
2019-11-05 Biking and walking to work in the US (and Sweden)
Data: https://github.com/rfordatascience/tidytuesday/tree/master/data/2019/2019-11-05
My code: https://github.com/mrtnj/rstuff/blob/master/tidytuesday/commute.R
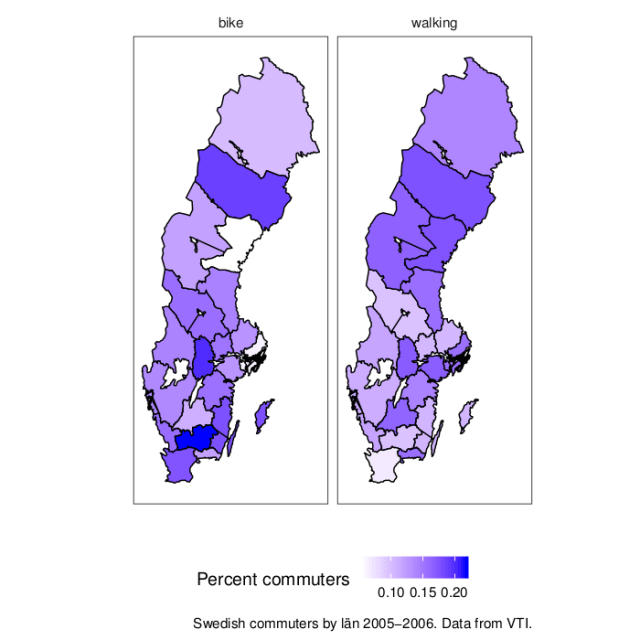
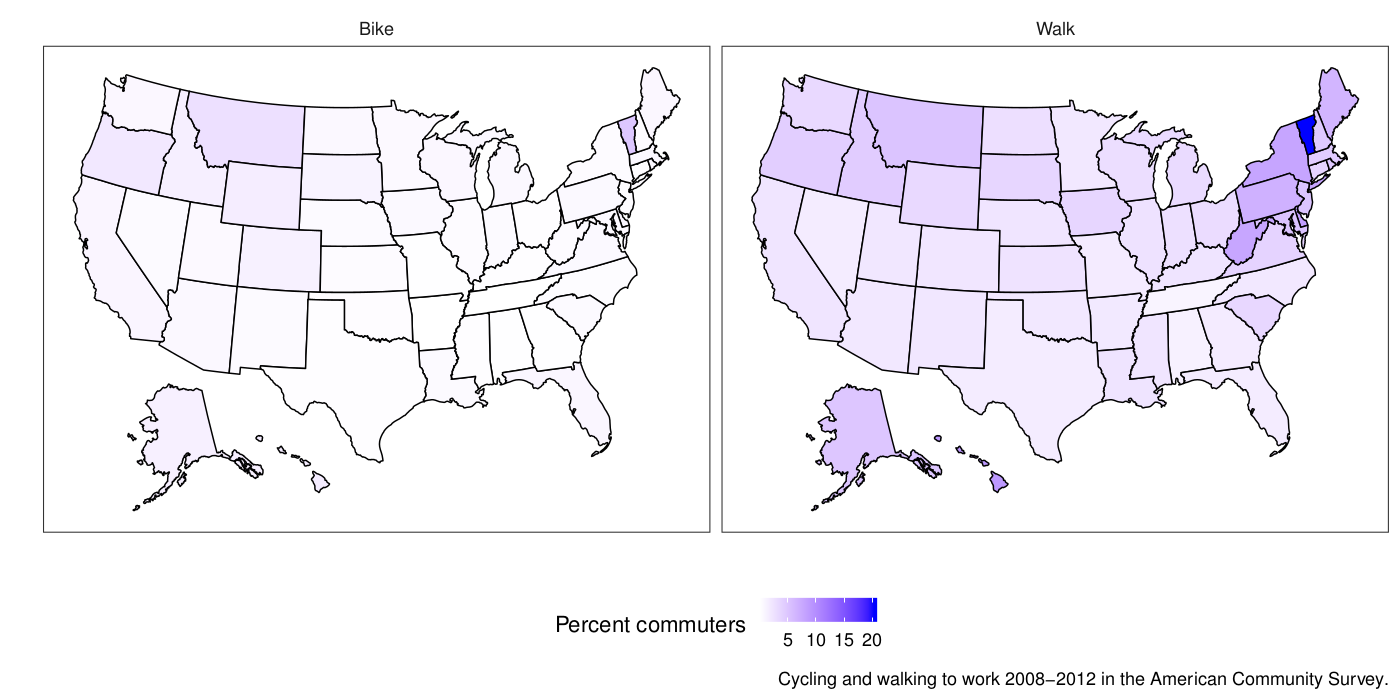
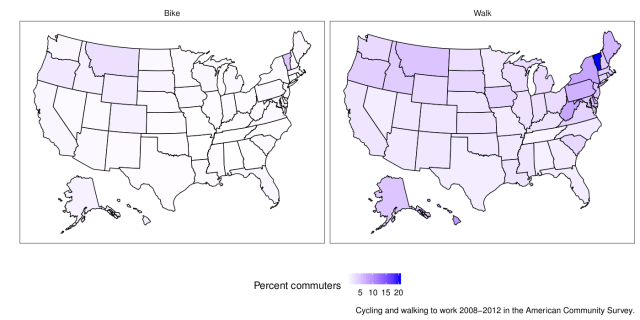
This week I felt I had to make a map. The end result doesn’t look like much, but it took a while. Here are the average percentages of commuters who walk and bike to work in different US states 2008-2012 with data from the American Community Survey:
library(dplyr)
library(ggplot2)
library(readr)
library(usmap)
commute <- read_csv("commute.csv")
## Map data from the usmap package
state_map <- us_map(regions = "state")
## There are some incompletely labelled states; fix them
missing <- setdiff(commute$state, state_map$full)
commute$state_modified <- commute$state
commute$state_modified[commute$state == "Ca"] <- "California"
commute$state_modified[commute$state == "Massachusett"] <- "Massachusetts"
We get map coordinates for the US states from the usmap package (because the one in maps doesn’t have Alaska and Hawaii).
Then we fix some mislabelling in the data.
## Get the average per state
state_average <- summarise(group_by(commute, state_modified, mode),
average = sum(percent * n)/sum(n))
## Combine averages and coordinates
combined <- inner_join(state_average,
state_map,
by = c("state_modified" = "full"))
We take a weighted average of the percentages per state and join the state averages with the state map coordinates. The map I posted on Twitter didn’t weight the average, but I think that is a bit better. There is still the issue that states have different populations and different distributions of large and small cities, but that’s the nature of things. In summary, there is not much biking going on, but some more walking to work.
plot_map <- ggplot() +
geom_polygon(aes(x = x, y = y, fill = average, group = group),
colour = "black",
data = combined) +
facet_wrap(~ mode) +
scale_fill_continuous(low = "white",
high = "blue",
name = "Percent commuters") +
theme_bw(base_size = 16) +
theme(panel.grid = element_blank(),
strip.background = element_blank(),
axis.text = element_blank(),
axis.ticks = element_blank(),
legend.position = "bottom") +
xlab("") +
ylab("") +
labs(caption = "Cycling and walking to work 2008-2012 in the American Community Survey.")
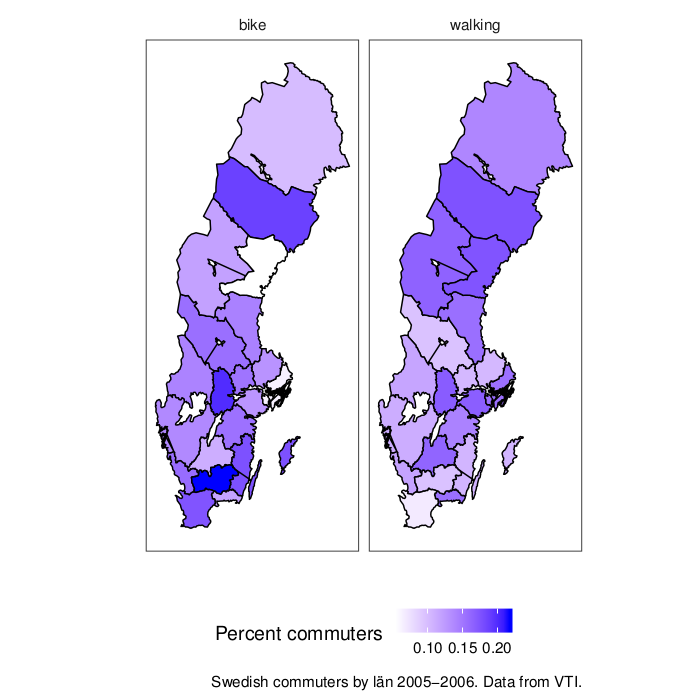
The US seems to live up to its reputation as a motorised country. But I have no feeling for the scale of the data. For comparision, here is a map of Sweden with some not too recent data (2005-2006, from this VTI report>). The map is from the swemap package.