SVT:s Vetenskapens värld sände nyligen (17 oktober, ”Ditt förutbestämda liv”) ett program om beteendegenetik. Det handlar om en studie i Dunedin, Nya Zeeland, som kopplar varianter av vissa gener, i kombination med påfrestande händelser under livets gång, till antisocialt beteende, depression med mera. SVT och dokumentären presenterar den som ”en oerhörd studie” som ”skrivit om den klassiska frågan om arv och miljö och visat att kombinationen är det avgörande”. Men det är snarare en stilstudie i hur välmenande forskare kan ha otur, dra förhastade slutsatser och skapa ett nystan av överdrifter. Otur och otur, förresten, för dem själva ledde det ju till berömmelse och dokumentärer som når ända till Sverige. Men för vetenskapen om den genetiska grunden för beteende var det ändå mest otur.
På 00-talet, när studierna ifråga publicerades, var beteendegenetiker väldigt optimistiska om vad som krävdes för att hitta gener som förklarar komplexa egenskaper, till exempel våldsamt beteende, depression med mera. Många trodde att det räckte att göra som i Dunedin, samla in data från kanske 1000 individer och välja ut en gen, en så kallad ”kandidatgen”, att studera. Komplexa egenskaper, som mänskligt beteende, kan visst ha en avsevärd ärftlig komponent. Men den består av hundratals, kanske tusentals, okända genetiska varianter med små effekter. Dagens genetik har gått vidare till att studera tiotusentals eller hundratusentals individer för att ha en chans att hitta några varianter, och till att studera alla gener samtidigt istället för att försöka gissa vilka kandidatgener som är viktiga.
Men tusen människor, hör jag er protestera, det är väl ändå många? I fallet MAOA tittar de bara på män, så där ryker hälften. Sedan är det ungefär en tredjedel av dem som har riskvarianten, och en bråkdel av dem som haft en dålig uppväxt. I dokumentären låter kopplingen mellan MAOA, dålig uppväxt och antisocialt beteende så övertygande. Richie Poulton, en av författarna, säger: ”Om man tittar på de killar som har riskvarianten av genen och som blev gravt illa behandlade, så uppvisade hela 85% av dem någon form av antisocialt beteende när de blivit vuxna [min översättning].” I själva verket, om man läser artikeln, så består gruppen han talar om – män med riskvarianten som blivit gravt illa behandlade under uppväxten – av 13 individer. De 85% han talar om är alltså elva män. Hur många av dem hade, enligt originalartikeln dömts för något våldsbrott vid 26 års ålder? Svaret är fyra. Med ett stickprov på 13 människor får man inga bra mått på vad riskvarianten har för effekt. Man får brus.
Och brus är precis vad som kommer ur kandidatgenstudier inom psykiatrisk genetik. Det går till ungefär så här: Någon hittar en kandidatgen i en liten studie, dåförtiden med stor buller och bång. Sedan kommer dussintals liknande studier med motsägelsefulla resultat. Ibland hittar de något liknande, ibland inte. Ibland hittar de en effekt på något annat: en interaktion med något nytt, en annan vagt relaterad egenskap. Efter hand börjar folk göra meta-analyser, som lägger ihop resultaten från många studier. De visar på stor variation och små effekter. Och så går det vidare. När det till slut börjar komma studier med större urval, som tittar på hela arvsmassan, så syns det (med några lysande undantag som apolipoprotein E) oftast inte ett spår av kandidatgenerna.
Men visst, ingen har tittat efter varianter i hela genomet med just de gen–miljöinteraktioner som var i Dunedinstudien. Och associationsstudier av hela genomet har hittills bara hittat varianter som kan förklara en bråkdel av den genetiska variationen. Så de gamla kandidatgensfavoriterna kanske också gömmer sig där ute, även om det inte ser ut så. Oavsett är det klart att de inte kan vara mer än en bråkdel av förklaringen, och att metoden att gissa kandidatgener och testa dem i små stickprov inte fungerar något vidare. Men på teve och i pressmeddelanden finns det aldrig komplikationer eller negativa resultat. Därför är MAOA också känd som ”the warrior gene”. Den är ett perfekt provokativt exempel att ta upp när man vill säga att människor är stenhårt programmerade av evolutionen att bete sig på ett visst sätt. Eller, som i den här dokumentären, när man vill komma ett mer humanistiskt budskap om hur uppväxten kan övervinna generna.
Författarna och dokumentärmakarna har såklart rätt i att både arv och miljö spelar roll för komplexa egenskaper. De har kanske till och med rätt att gen–miljöinteraktioner, där effekten av en viss genetisk variant bara visar sig under speciella miljöförhållanden, är viktiga. Men de har fel i att varianter i MAOA spelar en avgörande roll. Om MAOA-varianten har någon effekt alls, vilket inte ens är säkert, så är den bara en variant med liten effekt bland hundratals andra. Resultat som MAOA-associationen i Dunedin är inte några genombrott som skakar beteendegenetiken i grunden. De är ärliga misstag från en ung vetenskap som för 15 år sedan ännu inte hade lärt sig hur svårt det är att hitta gener som förklarar komplexa egenskaper. Istället för att älta dem är det dags att lämna kandidatgenerna bakom sig och gå vidare.
(Det här inlägget är lite försenat, för jag försökte få en kortare version av den här texten publicerad. Jag vet inte vad jag tänkte där. SVT Vetenskap har inte heller svarat. Den som läste den blev väl stött, eller avskrev den ungefär som en arg insändare. Nåja. Efter själva programmet var det ett par svenska forskare som fick vara med och prata lite. De var nog bra på sina ämnen, men ingen av dem verkade veta särskilt mycket om genetik, och sa inget kritiskt om själva innehållet. Ingen kritiserade heller det orimliga skrytet som dokumentären var full av. Jag förstår att jag framstår som en surgubbe nu, men det kan inte hjälpas.)
Litteratur
Caspi et al. (2002) ”Role of genotype in the cycle of violence in maltreated children.” Science
Caspi et al. (2003) ”Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene.” Science.
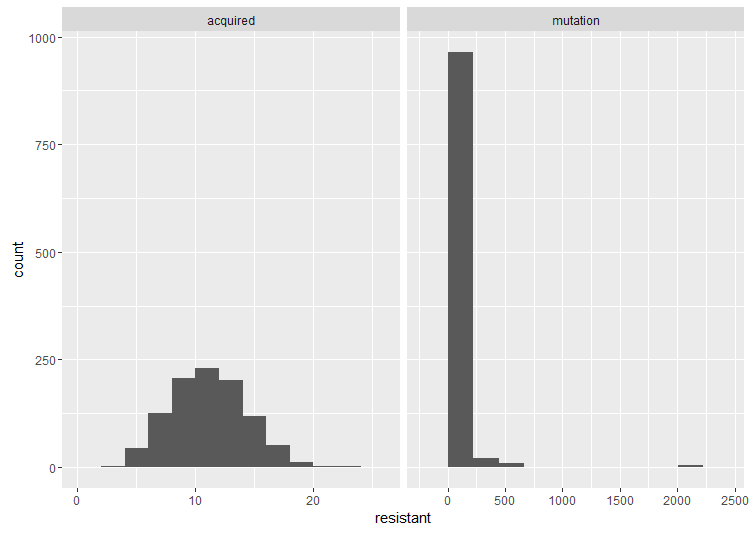
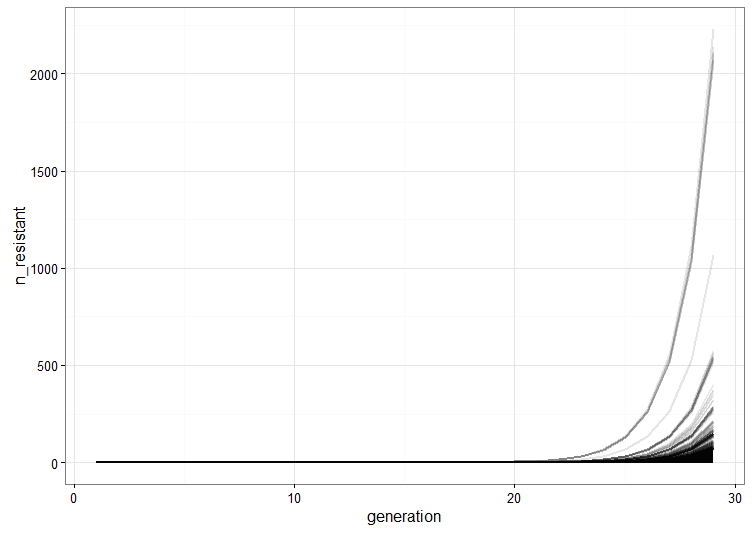
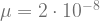 , which is quite close to their estimated value, and that the mutation can’t reverse. We also assume that the bacteria grow by doubling each generation up to 30 generations. We start a culture from a single susceptible bacterium, and let it grow for a number of generations before the phage is added. (We’re going to use discrete generations, while Luria & Delbrück use a continuous function.) Then:
, which is quite close to their estimated value, and that the mutation can’t reverse. We also assume that the bacteria grow by doubling each generation up to 30 generations. We start a culture from a single susceptible bacterium, and let it grow for a number of generations before the phage is added. (We’re going to use discrete generations, while Luria & Delbrück use a continuous function.) Then:

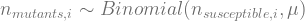 .
.
 .
.