Ärver blandrashundar samma ärftliga sjukdomar som de renrasiga hundar de härstammar ifrån?
Det är såklart svårt att säga något allmängiltigt. Hybrider, alltså korsningar av individer från olika populationer, kan nämligen bli väldigt annorlunda jämfört med sina föräldrar. I hybriderna träffas ju ibland genetiska varianter som vanligtvis inte brukar förekomma tillsammans i samma individ, och hybrider tenderar att bli heterozygota för många varianter. Så om varianterna ifråga råkar fungera så att heterozygoten får någon speciell egenskap eller att de interagerar med varandra på något sätt kan hybrider bli extrema på något sätt. Hybrider kan till exempel bli sjuka på något sätt (se ett exempel med hybridinkompatibilitet i gyckelblommor); om de blir särskilt starka och friska kallas det heteros.
När det gäller sjukdomsanlag beror det så klart på hur vanligt anlaget är i de olika populationer som blandrashundens föräldrar kommer ifrån. Om det är en sjukdomsvariant med enkelt dominant/recessivt arv så måste individen få anlaget från båda föräldrarna för att bli sjuk. Om det gäller en polygen sjukdom med många riskvarianter så minskar så klart risken ju färre av dem individen bär på. Så om det olika genetiska sjukdomar som är vanliga i olika raser, vilket verkar rimligt, så borde risken generellt bli mindre för blandrashundar än för rashundar.
Det var ungefär vad jag svarade då, om än väldigt mycket kortare, för jag var ganska trött. Så långt min spekulation: här kommer lite empiriska data! Bellumori & co (2013), Prevalence of inherited disorders among mixed-breed and purebreed dogs: 27,245 cases (1995-2010). Författarna har tittat veterinärjournaler från blandrashundar och rashundar som vårdats vid University of California-Davis Veterinary Medical Teaching Hospital och som haft olika sjukdomar med en genetisk komponent. Av 24 sjukdomar var det tio som var vanligare i renrasiga hundar, 13 ingen märkbar skillnad och en som var vanligare hos blandrashundar.
Sedan är det här en observationsstudie som kan påverkas av andra systematiska skillnader mellan hur många hundar som blir diagnostiserade grupperna än genetik. Till exempel är det inte orimligt att de som har en rashund kan vara mer på sin vakt efter sjukdomar som är vanliga i den rasen. Det kan också påverka resultaten av undersökningen.
Cytosinmetylering av dna är den klassiska molekylära epigenetiska mekanismen: alltså, någonting som inte ändrar dna-sekvensen men som ändå kan gå i arv: från modercell till dottercell vid celldelning och ibland till och mellan geneationerna vid sexuell reproduktion. Det som händer är att en av de fyra kvävebaserna i dna (cytosin, C) kan ha en extra metylgrupp eller inte. Metyleringsstatusen förs vidare när dna kopieras. Så, varför kallas cytosinmetylering inte en sekvensändring? Det ändrar bevisligen på dna-molekylens kemiska sammansättning. Jo, men det ändrar inte komplementariteten mellan baserna; C passar fortfarande med G och inte med de andra. Det ändrar inte heller på aminosyrasekvensen i kodande sekvenser när de skrivs av till rna och sedan används till proteinsyntes. Däremot kan de ändra hur andra proteiner binder till dna och på så sätt fungera genreglerande. Inte för inte tittar epigenetiska studier väldigt ofta på dna-metylering.
Epigenetik är intressant av flera anledningar: dels för att förstå hur celltyper i olika delar av organismen blir som de blir, dels för att det öppnar för intressanta transgenerationseffekter där saker som hänt föräldrarna eventuellt kan påverka avkomman och dels för den spännande tanken att dna-metylering skulle funka som ett extra lager av ärftlighet. Det skulle kunna fungera ungefär som genetik, men inte baserat på skillnader i dna-sekvens mellan individer utan på stabila skillnader i dna-metylering. Det finns några exempel, både hos djur och växter (Cubas m. fl. 1999), men de är en smula obskyra.
Häromveckan kom en artikel (Cortijo m.fl 2014) jag har väntat på sen i somras: den senaste i en serie experiment med en helt bisarr experimentpopulation som några galna (på ett bra sätt!) vetenskapare har kommit på. De har tagit fram en korsning av backtrav där alla individerna är genetiskt identiska (nästan helt) men har olika dna-metyleringsmönster. Detta därför att en av ursprungsväxterna i korsningen är en transgen planta som saknar en viktig gen som metylerar dna. Transgenen har de korsat ut, så avkomman har normal dna-metylering, men de har ändå ärvt olika metyleringsmönster från den ursprungsväxten. Det visar sig att flera egenskaper skiljer sig mellan individer med samma dna men olika dna-metylering, bland annat blomningstid och rotlängd. Det betyder att de egenskaperna kan påverkas av ärftliga dna-metyleringsskillnader. Även om de här skillnaderna är framtagna i labbet i en ganska artificiell situation visar det på att en skillnader i de här egenskaperna kan förklaras av epigenetik.
I den här artikeln har författarna gjort en metyleringsbaserad variant av genetisk kartläggning. De har alltså testat dna-metyleringen på regioner jämt utspridda i genomet (epigenetiska markörer!) och letat efter markörer associerade med egenskaperna. På så sätt hittar de kromosombitar som bör innehålla någon variant, i det här fallet en dna-metyleringsvariant, som orsakar en skillnad i egenskapen. Det är precis som genetisk kartläggning men med epigenetiska varianter istället för genetiska. Sedan får författarna precis samma svårigheter som en alltid får med genetisk kartläggning: de har associerade regioner på kromosomer. Vilken av alla gener i området är det som påverkats av en variant? Och, i det här fallet, vilken sekvens är det som är metylerad eller inte metylerad och får något att hända? Hur som helst kan de kartlägga de stabila epigenetiska varianter som kan förklara skillnader mellan individer i komplexa egenskaper som blomningstid. Nu börjar det likna något.
Okej, så det verkar som att associationen mellan intron 1 av FTO och övervikt samt diabetes förklaras av en reglerande effekt på granngenen, IRX3. Men vad gör IRX3 då? Hur är den inblandad i hur tung en blir? Ja, det är det ingen som riktigt vet. Först och främst har den en siffra i namnet, så alla kan gissa att det är den tredje i en familj av IRX-gener. Det är ganska typiskt för gener att de förkommer i familjer av liknande gener som bildats av duplicerande mutationer någon gång under den evolutionära historiens gång. De flesta djur har flera IRX-gener. Ryggradsdjur har sex stycken organiserade i två kluster på varsin kromosom. (Kerner m. fl. 2009) En bit efter FTO och IRX3 i människans kromsom 16 kommer IRX5 och sedan IRX6.
IRX står för Iroquois-familjen efter en muterad bananfluga vars borst tydligen ser ut som en tuppkamsfrisyr. De innehåller en homeodomän, ett återkommande motiv hos många proteiner som reglerar genuttryck. De har betydligt fler och intressantare funktioner än utvecklingen av borst: de är med och bygger upp kroppens former i flugembryon och nervsystemets och hjärtats utveckling hos ryggradsdjur. Sannolikt utövar familjen de funktionerna genom att reglera en väldigt massa andra gener, som transkriptionsfaktorer plägar göra. (Cavodeassi m. fl. 2001)
Så familjen är inblandad lite här och där och den uttrycks lite överallt. Titta på tabell 1 från Houweling m fl. (2001; artikeln är fritt tillgänglig och första tabellen kommer nästan direkt) som sammanfattar mätningar av genuttryck i olika delar av musembryon. IRX3 ligger i IrxB-klustret, så ett B i tabellen betyder att den uttrycks tillsammans med de andra i samma kluster. Etta A betyder är samma sak men för det andra klustret. Ett E betyder att den avviker från de andra två i klustret; ett I betyder att alla uttrycks lika mycket och ett streck att den litet eller inget uttryck. Det är flest A, B och I. Det vill säga: flera familjemedlemmar tenderar att uttryckas tillsammans, särskilt de i samma kluster och särskilt IRX3 och 5. Det här är inte heller så konstigt, men det gör det svårare att reda ut vad en enskild IRX-gen håller på med.
Som en liten illustration: möss utan IRX3 verkar enligt Smemo & co (2014; alltså den artikeln som föranledde den här serien poster) klara sig bra utan konstiga defekter, mer än att de är små och inte blir tjocka av fett foder. Men om hela IrxB-klustret tas bort (och tre andra gener, i och för sig, vilket naturligtvis kan vara en del av orsaken) blir resultatet stackars möss med diverse skelettdefekter (Peters m. fl. 2002). Både IRX3 och IRX5 verkar vara nödvändiga för hjärtat på olika sätt: IRX5 för att hjärtat ska uttrycka rätt jontransportprotiner (Costantini m. fl. 2005) och IRX3 för att det ska bilda cellkontakter och sprida nervsignalen när det ska slå (Zhang m. fl. 2011).
Efter det ovanstående verkar det ju inte så långsökt att Smemo & co tittar på IRX3-uttryck i hjärnan. Deras hypotes om hur IRX3 påverkar vikten är att den skulle mixtra med hjärnans signallering till fettvävnaden och öka förbränningen. Finns det något annat i litteraturen som knyter IRX3 till ämnesomsättning eller aptit? Ja! Redan 2010 kom nämligen en artikel som hävdade att FTO-association kanske förklarades av IRX3-reglering (Ragvin m. fl. 2010). Deras angreppssätt för att hitta reglerande regioner var inte som Smemo & co att fånga in kromosombitar som interagerar, utan att titta efter evolutionära mönster. Viktiga delar av genomet tenderar att konserveras därför att naturligt urval motverkar mutationer som ändrar deras funktion. Oviktiga delar, vilket är lejonparten av genomet, kan muteras sig i stort sett hur mycket som helst.
De hittade konserverande icke-kodande sekvenser nära FTO och testade dem i ett så kallat reporterexperiment, vilket betyder att en sätter in sekvensen i någon organism tillsammans med någon gen som är lätt att detektera när den uttrycks. I det här fallet använde de ett grönt fluorescerande protein (som heter GFP … väldigt fantasifullt) och zebrafiskembryon. Om den konserverade sekvensen verkligen är reglerande kommer cellerna alltså fluorescera grönt när de belyses med ljus av rätt våglängd. Mycket riktigt, de associerade varianterna ligger i reglerande sekvenser som är aktiva i delar av embryot där IRX3 också är aktivt, bland annat i bukspottkörteln.
Bukspottkörteln, ja. Alla diabetesintresserade borde höja på ögonbrynen nu. Författarna prövade att slå ut IRX3 i fiskembryon och fann att det påverkade bildningen av både insulin-, ghrelin- och glukagonproducerande celler. Alla tre är viktiga hormoner för ämnesomsättningen. Insulin och glukagon reglerar blodsocker och ghrelin reglerar aptit. Kort och gott: Smemo & co och och Ragvin & co har båda resultat som tyder på att det är IRX3 som är den viktiga genen. Men de föreslår olika mekanismer, och det kan mycket väl vara både och.
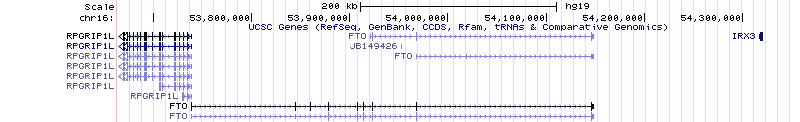
Detta har hänt: Hur mycket människor väger har en genetisk komponent och det finns flera studier som kopplar varianter i en gen som heter FTO till övervikt och typ 2-diabetes. Precis vilken den orsakande varianten är och hur den påverkar vikt är inte klart. Häromdagen publicerades en vetenskaplig artikel med resultat som tyder på att varianterna, även om ligger i FTO, kanske utövar sin effekt genom att påverka regleringen av en helt annan gen som ligger en bra bit bort, IRX3. Både FTO och IRX3 verkar ha effekter på vikt i experiment med genetiskt förändrade möss. Förvirringen om vad som egentligen pågår blir alltså ännu större, om än på en högre nivå. I fredags skrev jag lite om detta men utan att gå in på vad artikeln egentligen handlade om. I den här posten ska vi skruva upp genetiknördigheten en smula. Låt oss börja med en bild: så här ser området med FTO och IRX3 ut i UCSC-genomläsaren. Det är en bit av det mänskliga referengenomet, kromosom 16, med kända gener utritade.
Först och främst, vad är problemet egentligen? Det finns en association till varianter som ligger i FTO. De ändrar i och för sig inte på den kodande delen av genen, men de ligger i första intronen, där det rätt ofta finns reglerande sekvenser. (Titta på spåren märkta ”FTO” i bilden ovan. De kodande bitarna är de tjockare lådorna och intronerna är strecket emellan. IRX3 är nästa gen längs kromosomen.) FTO är den uppenbara kandidaten. Tidigare har folk använt två sorters experiment för att pröva om FTO faktiskt är den orsakande genen och de har fått resultat som förefaller motsäga varandra. Å ena sidan, att mixtra med genen i möss. Det är ett sätt att titta på genens normala funktion: om mössen ökar eller minskar i vikt i jämförelse med kontrollmöss har den antagligen med viktreglering att göra … på något sätt. Och mycket riktigt: möss utan FTO blir magra och möss som uttrycker extra mycket FTO blir stora.
Å andra sidan, genuttryckskartläggning. Det vill säga: Om de genetiska varianterna verkligen har en reglerande effekt borde uttryck av FTO, alltså hur mycket av genen som tillverkas, också vara associerat med samma varianter. Men så är det inte. Så även om FTO visst är inblandat i vikt på något sätt, så verkar det inte vara den underliggande genen till associationen i människor. Om inte det viktiga händer i någon vävnad vid någon tidpunkt där ingen ännu tittat, vill säga.
Hur får en då veta om varianterna kanske reglerar någon annan gen? Ett sätt är att leta efter vilka delar av dna-strängen som är fysiskt nära varandra i cellkärnan. Det där kan behöva en förklaring. Vanligtvis när jag skriver att sekvenser är ”nära” varandra menar avståndet längs dna-strängen. Men när kromosomen är i sitt verkliga tillstånd i cellkärnan ligger den delvis ihoplindad, delvis utsträckt och reglerande sekvenser som påverkar varandra är också nära varandra i rymden. Den teknik författarna använt, circular chromosome conformation capture, går ut på att fånga in sådana sekvensbitar som rör vid varandra, sekvensera dem och på så sätt bygga upp en karta över vilka kromosombitar som har reglerande interaktioner. Det är förstås inte självklart att två bitar som råkar vara nära varandra har någon sorts reglerande interaktion, men om de förekommer tillsammans tillräckligt pekar det i alla fall i den riktningen.
De undersökte den del av FTO-genen som är associerad med övervikt i människor i vävnadsprover från möss. Det visar sig den FTO-biten (47 000 baser) ofta befinner sig nära inte bara området före själva FTO-genen, vilket sannolikt innehåller genens viktigaste reglerande sekvens (promotorn), utan också med IRX3, som ligger en ganska bra bit bort. Och när de sedan tog fram genetiskt förändrade IRX3-knockout-möss visade de sig väga mindre och när de sattes på högfettdiet gå upp mindre i vikt och bli mindre insulinresistenta än vanliga möss. Det är de här genetiskt förändrade mössen som en av författarna, Chin-Chung Hui, beskrev som ”helt resistenta mot fetma orsakad av fet mat” (TT). Dessutom, att mixtra med IRX3 verkar inte ha någon effekt på uttrycket av FTO. Den förefaller verka oberoende av FTO.
Så långt mössen! Författarna tittade på genuttryck i mänsklig hjärna: är varianterna som kopplats till övervikt också associerade med genuttryck? Som förut, ingen association med uttryck av FTO, men med IRX3! Effekten är inte överväldigande tydlig, men det tyder i alla fall på att varianterna i FTO faktiskt har en reglerande effekt på IRX3.
Vart leder allt det här? Sammantaget verkar IRX3 vara en bättre kandidat till att vara den orsakande genen än FTO. Även om tidigare resultat ganska klart visar att FTO också har något med vikt att göra, så verkar det som att just den här varianten, även om den ligger i en intron av FTO faktiskt utövar sin effekt genom att reglera en annan gen. Så rörigt kan det vara.
”Fetma-gen upptäckt. Man blir aldrig fet om man saknar den”, stod det. Och jag tänkte: jag är tvungen att blogga om det här, eller hur? Det handlar om genen FTO, som sedan tidigare är känd för att vara kopplad till övervikt i associationsstudier. Det vill säga: vissa människor har en variant av FTO som gör dem, i medeltal, tyngre än de som har en annan FTO-variant. FTO är absolut inte den enda förklaringen till ärftliga skillnader i vikt, men den har en hyfsat stor effekt, tydligen i medeltal 3 kg skillnad mellan homozygoterna. Frågan är bara hur det går till? Vad sjutton är det genen gör, och vad är det för skillnad på varianten som gör en tyngre och den som gör en lättare? En ny artikel (Smemo m.fl 2014) tyder på att varianterna, även om de ligger i FTO, kanske utövar sin effekt genom att reglera en helt annan gen, IRX3. TT:s text av Johan Nilsson (DN, DI, SvD) är inte så illa:
Upptäckten, som presenteras i tidskriften Nature, visar samtidigt hur komplicerad arvsmassans reglering kan vara. /…/ Forskare från flera länder började då undersöka saken närmare och har nu upptäckt att små delar inuti FTO-genen i själva verket styr en helt annan gen: IRX3, som ligger hundratusentals baspar bort från FTO-genen.
Det är ofta bra att tänka sig två typer av genetik: Genetik i den första bemärkelsen (obs, numreringen är min egen och inte tillämplig i något annat sammanhang) handlar om molekylära gener och deras funktion utan vidare hänsyn till genetisk variation. Det är den typen av data en får sig till livs genom att slå upp gener i de flesta databaser, en beskrivning av proteinet som den kodar för och vilka typer av reaktioner eller processer det deltar i. Det är den typen av information som genernas jobbiga akronymer till namn ofta står för. Det är sådant en får veta genom att slå ut en gen i någon försöksorganism och se vilka processer som inte fungerar utan den. Genetik i den andra bemärkelsen handlar om genetisk variation: när en del individer har en viss variant av en gen, andra individer har en annan, och de varianterna fungerar olika. Det är den här typen av genetik som förklarar ärftliga skillnader mellan individer och populationer och den typ av data som kommer ur genetisk kartläggning. Det handlar naturligtvis också om geners funktion, men mer specifikt hur genetiska varianter ändrar geners funktion.
Den här artikeln kombinerar experiment faller i båda kategorierna. Syftet är att förklara hur genetiska varianter i FTO som upptäckts i associationsstudier fungerar, vilket är genetik i den andra bemärkelsen. Men för att göra det använder de bland annat genetiskt modifierade möss utan IRX3, och det är ett experiment i den första kategorin. När det står så här (TT-artikeln) handlar det alltså om genetiskt förändrade knock-out-möss:
Möss som saknar genen förblir smala, nästan oavsett hur mycket fet mat de äter. Dessutom tycks de inte utveckla diabetes.
Det är alltså inte en beskrivning av något en kan och bör göra i människor som någon sorts bantningskur. Det är utfallet i ett experiment som visar att IRX3 har något med viktreglering att göra. Okej, men vad har den med viktreglering att göra och hur hänger de två generna FTO och IRX3 ihop med varandra? Artikeln ifråga innehåller imponerande experiment om just detta. Den otåliga läsaren kan titta vidare på själva artikeln, men jag tänkte återkomma om några dagar med en sammanfattning.
Någon frågade på Facebook ungefär såhär: Fåglarna har andra könskromosomer än vi, ZW istället för XY, och det är honorna som har den lilla konstiga kromosomen och hanarna som har två av samma (Z). Varför kallas ZW-individen hona och inte hane, när XY-däggdjur kallas hanar?
Jo, för att kön (nu i den strikt biologiska bemärkelsen) inte handlar om kromsomer eller ens hur olika delar av kroppen ser ut; det handlar bara om könsceller. Den som gör de stora könscellerna är hona och den som gör de små är hane. Det finns arter som har sexuell reproduktion men där alla producerar lika stora könsceller, men när det gäller arter som har flera storlekar är det oftast två. Den delen av kön är en dikotomi, men sedan blir det intressantare. Han- och honfunktionen kan sitta på olika kroppar eller samma; de kan komma i olika former och könsbestämningen kan vara genetisk, styras av någon miljöfaktor, vara bestämd från konceptionen eller ändras under livets gång. Här är en schyst liten video från Aaron Reedy och TED Ed (och som jag såg först på Melissa Wilson Sayres blogg) om könsbestämning i olika djur. Genetisk könsbestämning med könskromosomer är inte allmängiltig!
Videon innehåller några små konstigheter, som den enkelsträngade dna-molekylen i den haploida myran, men låt oss inte gräva ner oss i det. Och naturligtvis betyder ”pojke” och ”flicka” betydligt mer än sekundära könskaraktäristika. Förresten, hitta Nemo!
The distinction between ”gene” in the sense of an allele at some locus and ”gene” in the sense of a dna sequence with a name and some function seems easy enough, but still causes a lot of confusion, both in popular and scientific literature.
This was very clear a few months ago when science journalist David Dobbs published his ”Die selfish gene, die” and a few weeks of debate broke out. In my opinion it’s not a particularly good piece, but I agree with Dobbs that the ”selfish gene” metaphor sometimes invites misunderstandings. The article itself displays a few of them, when it suggests that evolution and genetics as understood before the age of microarrays are somehow at odds with the importance of gene regulation or phenotypic plasticity. I suspect that many of these problems stem from the double meaning of the word gene. Other examples are found in headlines claiming that researchers have found the gene for something or the confusion about the word pleiotropy (Paaby & Rockman 2012).
When Dawkins wrote about the selfish gene, he did not mean the selfish dna sequence encoding a protein; he meant the selfish genetic variant causing differences in fitness between individuals. (Or rather, a set of genetic variants in sufficiently close linkage to seldom be separated by recombination.) The book is not about molecular genes. As anyone who actually read it knows, it deals mostly with behaviour using game theory approaches. This does not mean that Dawkins denied that there are actual molecular genes doing the mechanistic work, but that he analysed the situation mostly on a different level. And had he chosen to write only about known sequence variants with adaptive effects on behaviour it would have been a very short book.
Of course the word ”selfish”, while I agree that it is the proper word in the sense that Dawkins intended, is great for those who want to point to instances where people are horrible to each other and tell you that it’s all because of evolution. But I think that is a bigger issue that will not be solved by tweaking popular science metaphors. By the way, that is completely contrary to Dawkins’ intentions, which were to popularise the evolutionary models that explain why animals are not always horrible to each other, even though their behaviour is shaped by natural selection.
Ännu en video från ESEB2013 i somras! Luisa Pallares berättar om genetisk kartläggning, med helgenomsassociation, av skallens form hos naturliga hybrider mellan två underarter av möss. Bara för att möss och bananflugor finns som inavlade labbstammar ska en inte glömma att de har livaktiga genpooler i naturen med intressant variation. Det klassiska sättet, om jag vågar kalla det så, att kartlägga genetiska varianter som skiljer två populationer är att göra experimentella korsningar mellan dem — jag jobbar till exempel på en korsning mellan tamhöns och röda djungelhöns. Men när de tittar naturliga hybrider får de både den höga upplösningen i en associationsstudie och styrkan i att jämföra två olika populationer.
(Bonus: Hitta min kalufs i publiken. Ledtråd: Jag sitter bredvid min handledare, och om en tar medelvärdet av längden på vårt hår blir det ungefär en pottfrisyr.)
This recent paper, Pandey & al (2014), made me interested because I’m in the business of finding genes for traits, and have spent quite some time looking at lists of gene names and annotation database output. One is tempted to look for the ”outstanding candidates” that ”make biological sense” (quotes intended as scare quotes), but the truth is probably that no-one knows what genes and functions we should expect to be affected by genetic variation in, for instance, behaviour. This paper tries to make the case for the unknown parts of the brain transcriptome; they use data about gene expression, protein domains, paralogs and literature to argue that the unknown genes are unknown for no good reason and that they might be just as important as genes that happen to be well-known.
They found genes that are had a high ratio of expression in brain to average expression in other tissues of C57BL/6J and DBA/2J mice and searched PubMed for these genes in combination with neuroscience-related keywords. Some of them have few citations and these are their selectively expressed but little studied genes. They then make a series of comparisons between these and well-studied genes. It turns out the only major difference is that well-studied genes were discovered (entered into GenBank) earlier.
Comments:
I don’t know to what extent these results are suprising. I was not surprised by their main conclusion, but then again, that maybe my opinion was mostly prejudice. There is a literature on biases in the functional genomics literature, but I don’t know much about it. And apparently neither did the authors, initially, as Robert Williams writes in a comment on the PLOS ONE website:
We did not rediscover the lovely work of Robert Hoffmann (now head of WikiGene) until the paper had been submitted in succession to six higher profile journals … Hoffmann and colleagues showed that social factors account for much of the annotation imbalance for genes.
I love the idea of authors writing an informal comment about the background of the paper like this.
The coexpression network results show some of the little known genes are just as connected as known important genes. This suggest some of the unknown genes might be important too, if we can trust that coexpression hub genes are likely to be important (for various values of ”important”). Maybe this is a scientific opportunity for some neuroscientist. Several people I’ve talked with has imagined future Big Science initiatives to describe the function of unknown genes — ”divide them up between labs and characterise them!” — and some initiatives exist, such as the IMPC. On the other hand, how do we know that we really find the most important and interesting functions of a gene? The skeptic in me thinks that going bottom up, from gene to phenotype, will miss the most interesting surprising phenotypes.
I think ”ignorome” is one of those unnecessary bad omics words, which is why I’ve avoided using it.
Their PubMed query was restricted to mouse, human and rat. I wonder why. Maybe there could be something useful from fruit flies or roundworms?
Overall, a fun paper that I recommend reading over a few cups of coffee!
A couple of weeks ago I attended the Evolution in Sweden meeting in Uppsala, as expected a very nice meeting with lots of interesting things. My last conference was ESEB last summer, which was great because it was a huge conference with so much to see and so many people. Evolution in Sweden was great because it wasn’t huge, so that it was very possible to see everything, recognise familiar faces and talk with people. I had a poster on the behaviour genetics of chicken domestication (of course!).
Here are some of my personal highlights, in no particular order:
Kerstin Johannesson’s talk, an ”advertisement for marine organsims” was probably the most fun and engaging. I was very convinced that evolutionary research in the Baltic Sea is a great idea! Among other things she mentioned salinity gradients, the sexual and asexual reproduction of Fucus brown algae, Littorinasaxatilis of course and the IMAGO project to sequence and assemble reference genomes for eight different species from the Baltic.
We have a great infrastructure for evolutionary research: the Baltic Sea. [quoted from memory]
Claudia Köhler spoke about why triploids in Arabidopsis thaliana fail, which is an interesting story involving the endosperm, which in a triploid seed turns out tetraploid, and genomic imprinting. They screened for mutants able to form triploid seeds and found paternally imprinted gene, that is dosage-sensitive and causes the failure of triploid seeds (Kradolfer & al 2013).
Anna Qvarnström and Hans Ellegren talked about different flycatcher projects. I don’t have that much clever to say about this right now, except that both projects are really fascinating and impressive. Everyone who cares about genomics in the wild should keep an eye on this.
There were two talks from Umeå Plant Science Centre: Stefan Jansson’s about association mapping in aspen (SwAsp), which sounds fun but difficult with tons of genetic variation, and Pär K. Ingvarsson’s about the Norway spruce genome (Nystedt & al 2013). An interesting observation from the latter was that it’s gigantic genome size (~20 Gb) apparently isn’t due to whole-genome duplications, but to unchecked transposable element activity. A nice nugget to remember: about half of the sequence, or three to four human genomes, consists of LTR-type repeats.
I’m afraid you will never read very much from me about theory talks. I am an engineer after all, so I don’t fear the equations that much, but most of the time I don’t have necessary context to have any clue where this particular model fits into the grand scheme of things. However, Jessica Abbott gave a fun talk presenting a model for sexual conflict in hermaphrodites that deserves a special mention.
I did see quite few a genomic plots of Fst outliers and I believe the question that needs answering about them is: What do they really mean? One can do comparisons of comparisons (like in Roger Butlin’s talk and their paper on parallel evolution of morphs in Littorina; Butlin & al 2013), but when it comes to picking out the most differentiated loci on a genome-wide level, are they really the most interesting loci? Are the loci of highest differentiation the loci of adaptation; are they the loci of speciation? (Ellegren’s talk and the flycatcher genome paper; Ellegren & al 2012). It’s a bit like the problem faced by QTL mappers — ”now that we’ve got a few genomic regions, what do we do with them?” — with the added complication that we don’t have a phenotype associated with them.
Genetic architecture wasn’t an explicit theme of the meeting, but it always comes up, doesn’t it? Will traits be massively polygenic, dooming researchers to a lifetime search for missing heritability, or relatively simple with a handful of loci? And under what circumstances will either architecture occur? Jon Ågren talked about the fantastic Arabidopsis thaliana in situ QTL mapping experiment. I think it is best illustrated with the video he showed last time I heard him talk about this — Lost in transplantation:
Folmer Bokma used Lego dinosaurs to great effect to illustrate developmental constraints. Also a large part of the talk was quotes from different famous evolutionary biologists. Very memorable, but I’m not sure I understood where he was heading. I was expecting him to start talking about the need for G matrix methods any moment. My lack of understanding is of course my fault as well, not just of the speaker’s, and there were a few graphs of gene duplications and gene expression data in primates, but I don’t feel that he showed ”how phylogenetic analyses of genomic data can shed new light on these ideas”, as promised in the abstract.
Possibly the best expression of the meeting: Erik Svensson’s ”next generation fieldwork”. I’m not a fan of the inflation of words ending in -omics (and I sometimes feel ”genomics” should just be ”genetics”), but if we have genomics and proteomics, phenomics is also justified, I guess. As a tounge-in-cheek version ”next generation fieldwork” is spot on. And very true: clever phenotyping strategies in natural populations and natural settings is more even more important than rapid sequencing and genotyping. By the way, Erik Svensson, Jessica Abbott, Maren Wellenreuther and their groups have a lab blog which seems nice and active.
And finally, the thing that wasn’t so great, coincidentally, the same thing that wasn’t so great at ESEB: the gender balance: only 7 out of 28 speakers were women. I don’t know to what extent that ratio reflect the gender ratio of Swedish evolutionary biology, but regardless it is too low.
It’s been a while since mid-January, but I’ve been busy (with some fun things — will tell you more later). And maybe we’ll see each other at the next Evolution in Sweden in Lund.






{kind=link}